There is a growing concern about accelerated aging among the rapidly increasing number of cancer survivors. Clinically, “accelerated aging” phenotypes in cancer survivors include premature mortality and comorbidities – secondary cancers, frailty, chronic organ dysfunction, and cognitive impairment which can impact long-term health and quality of life in cancer survivors.
- accelerated aging
- cancer treatment
- cellular senescence
1. Introduction
Individuals with and without cancer can age very differently from one another. People with cancer seem to age very rapidly, may appear frail due to cancer treatment, and need assistance in daily routines at age 70, whereas individuals without cancer may not need assistance even at ages much older than 70 [1]. Growing evidence demonstrates that individuals with cancer age faster, so their biological age appears to be older than their chronological age (so-called accelerated aging). According to the definition proposed by Baker and Sprott [2], biological age is characterized by the biological parameter[s] of an organism, either alone or in some multivariate composite. Clinically, “accelerated aging” phenotypes in cancer survivors are characterized by the development of age-related health conditions, including premature mortality and comorbidities – secondary cancers, frailty, chronic organ dysfunction, and cognitive impairment which can impact long-term health and quality of life in cancer survivors. Unfortunately, many therapies used to treat or control cancer may lead to unintended consequences that appear to accelerate aging process [3,4,5]. Cancer treatments can lead to accelerated aging by inciting hallmarks of aging, including telomere attrition, stem cell exhaustion, cellular senescence, DNA damage, and epigenetic alterations [6,7].
2. Cancer Treatment-Induced Accelerated Aging in Cancer Survivors
It is not uncommon in clinical practice to observe that cancer patients receiving treatments appear to become “older”. For example, as discussed in Hill et al. [8], a healthy 65-year-old female who “appears to be age 65” is diagnosed with locally advanced breast cancer and receives doxorubicin, cyclophosphamide, and paclitaxel. By the time she completes treatment, she becomes frail and “appears older than age 65” [8]. After stopping the treatment, the acceleration of aging may slow down, sometimes may even reverse, but this patient may not “appear to be age 65” like how she appears to be at the pretreatment level (Figure 1) [8].

Figure 1. Hypothesized accelerated aging due to cancer treatments, adapted with permission from Guida et al. (2019) [9].
2.1. Epidemiological Evidence for Treatment-Induced Accelerated Aging
A growing body of literature is pointing toward the effects of cancer treatments on accelerated aging in cancer survivors.
2.1.1. Increased Frailty in Cancer Survivors
There is no one “tool” that is used in the clinical setting to assess frailty, but rather a variety of measures encompassing physical strength, nutritional status, cognition, and physical performance. In research, accelerated aging in cancer survivors has been captured by using some of these measurements such as grip strength, the timed up-and-go (TUG), and the 6-min walk (6MW) test [10]. Lintermans et al. conducted two studies comparing the grip strength after 6 months and 12 months of therapy to the baseline in women with breast cancer receiving aromatase inhibitors, and both studies found a decrease in grip strength after receiving treatment [11,12]. For example, after 6 months of therapy, they found that the grip strength decreased by 8% and 11% for left hand and right hand, respectively (p = 0.009) [11]. Furthermore, the Childhood Cancer Survivor Study (CCSS) assessed physical function in young childhood cancer survivors (n = 183 cancer survivors, who survived for at least five years; mean age (SD) = 13.5 (2.5)) and their age- and sex-matched siblings (n = 147; mean age (SD) = 13.4 (2.4)). This study showed that cancer survivors performed worse than their matched siblings on TUG (p = 0.003), and 6 MW (p = 0.002), but not grip strength tests (p = 0.49) [10]. In agreement with the results from the CCSS, the Rabin Medical Center study (n = 26) also reported a worse performance of 6MW test in allogeneic hematopoietic cell transplantation (HCT) survivors compared to their age-matched healthy controls (p < 0.005). That study also found no difference in hand grip in HCT survivors and their age-matched controls [13].
Another example stems from studies of hematopoietic cell transplantation (HCT), which reported an increased frailty in cancer survivors [14]. The Bone Marrow Transplant Survivor study (BMTSS) compared risk of frailty in 998 young adult HCT survivors (ages 18–64), who had received HCT for hematologic malignant diseases or severe aplastic anemia and survived at least two years after HCT, and 297 matched siblings (ages 18–64). The study reported that HCT recipients had a higher prevalence of frailty (8.4%) compared to their matched siblings (0.2%) (p< 0.001) [14]. The high prevalence of frailty in HCT recipients may be related to their exposure to the high-intensity chemotherapy, radiation, and immunosuppressive agents before, during, and after HCT. These high intensity treatments and complications injure normal tissue and may increase the risk of frailty after HCT, even among non-geriatric HCT patients [14].
2.1.2. Increased Risk of Comorbidities and Premature Mortality in Cancer Survivors
Previous studies suggested that cancer treatments led to accelerated aging that manifested as an increased risk of secondary neoplasms in cancer survivors. For example, another CCSS study of 14,359 childhood cancer survivors, who survived for at least five years (age range: 5–56 years) reported that, after childhood cancer diagnosis (maximum follow-up was 30 years), the cumulative cancer incidence of cancer was 20.5% (95% confidence interval (CI): 19.1–21.8%) for first occurrence of subsequent neoplasms and 7.9% (95% CI: 7.2–8.5%) for second malignant neoplasms (excluding non-melanoma skin cancer). The greatest cumulative incidence was among people surviving Hodgkin lymphoma (HL). This study also found a 170% (95% CI: 2.2–3.3) higher risk of all first subsequent neoplasms in cancer survivors who had been exposed to radiation therapy for their primary cancer diagnosed before 21 years of age compared to those who had not been exposed to radiation therapy [15]. Results from this study suggested that cancer survivors were at a higher risk of developing secondary neoplasm, and diagnosis of HL and treatment with radiation therapy were associated with an increased risk of secondary neoplasm [15].
Other studies reported an increased risk of cardiovascular diseases (CVDs) in both childhood and adult cancer survivors after treatment. For example, the St. Jude lifetime cohort study compared the cumulative incidence CVDs in 670 pediatric, adolescent, or young adult HL survivors with their age- and sex-matched controls from general population (n = 270), who were selected regardless of past medical history [16]. That study showed that the incidence of at least one adverse event of grade 3–5 involving chronic cardiovascular health conditions by age 50 was 45.5% (95% CI: 36.6%–54.3%) for the HL survivors, who had been treated with chemotherapeutic agents and radiation dosimetry compared to a cumulative incidence of 15.7% (95% CI: 7.0%–24.4%) in their matched controls [16]. Furthermore, the Kaiser Permanente Southern California study (KPSC) (36,232 over two-year adult cancer survivors and 73,545 matched cancer-free KPSC members) reported a higher incidence rate of CVDs in breast (incidence rate ratio (IRR)= 1.13, 95% CI= 1.06–1.22), kidney (IRR = 1.24, 95%CI: 1.02–1.51), lung (IRR = 1.58, 95% CI: 1.30–1.90), and ovary (IRR= 1.41, 95% CI: 1.06–1.88) cancer survivors compared to their age-, sex-, and residual zip code-matched cancer-free controls [17]. This study also reported an 11% decreased incidence rate of CVDs in prostate cancer survivors compared to their matched controls (95% CI: 0.84–0.95). However, that study found no difference in incidence rate of CVD in overall cancer survivors, and survivors of bladder, chronic lymphocytic leukemia, colon, rectal, thyroid, and uterus cancers compared to their matched controls [17]. Another example stems from the UK electronic health records databases study [18]. This study reported an increased risk of venous thromboembolism in cancer survivors of colorectum, lung, breast, uterus, prostate, and non-HL who were treated with chemotherapy compared to survivors of these cancers who did not receive chemotherapy [18].
Another CCSS study (n = 6,148) assessed the increased risk of late mortality (more than five years after diagnosis) in long-term survivors of childhood acute lymphoblastic leukemia (ALL) as a result of their cancer treatment [19]. With a maximum of 20 years follow-up, the overall late mortality rate was 360% (95% CI: 4.2–5.1) higher than the rate in their age-, sex-, and race-matched US population [19]. Furthermore, Yeh et al. developed a model to estimate the life expectancy for a cohort of 15-year-old five years ALL survivors [20]. The estimated life expectancy for these ALL survivors was 50.6 years, which equals to a loss of 10.4 years compared to the general population [20].
Literature pointing to the accelerated aging in people with cancer syndromes is scarce. Previous studies hypothesized that people with Li Fraumeni syndrome (LFS) were in the trajectory of accelerated aging, as people with LFS were at a greater risk of developing cancer [21]. In addition, results from studies showed that people with hereditary syndromes like Fanconi anemia were more susceptible to damage from cancer therapies such as radiation treatment [22].
These data collectively demonstrated that cancer survivors had lower physical function, higher risks of premature death, and age-associated morbidities such as secondary neoplasms and CVDs, suggesting an accelerated aging process after treatment.
2.2. Biological Mechanisms Underlying Treatment-Induced Accelerated Aging in Cancer Survivors
The potential biological mechanisms (Figure 2.) that explain how cancer treatments may incite hallmarks of aging (telomere attrition, stem cell exhaustion, cellular senescence, DNA damage, and epigenetic alterations) are shown in Figure 2 [6].
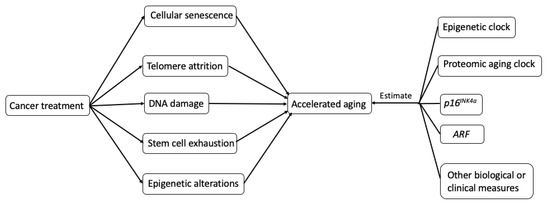
Figure 2. Possible biological mechanisms and potential measurements of treatment-induced accelerating aging in cancer survivors.
2.2.1. Cellular Senescence and SASP
Cellular Senescence and Aging
Cellular senescence is defined as an irreversible arrest of cell proliferation. Cellular senescence was hypothesized to be a tumor-suppressing mechanism [23]. Based on the hypothesis that cancer cells can proliferate indefinitely, cellular senescence can preclude cancer cells from proliferating. Since it may preclude cancer cells from proliferating, cellular senescence has been seen as a powerful strategy for cancer treatment. Many commonly used cancer treatments—e.g., chemotherapy, radiotherapy, CDK4/6 inhibitors, epigenetic modulators, and immunotherapy—intend to induce senescence in tumor cells. However, the same therapies also cause cellular senescence in neighboring non-tumor tissues [24]. Also, cellular senescence is often accompanied by a complex pro-inflammatory response—senescence-associated secretory phenotype (SASP). SASP is characterized by the secretion of SASP factors including numerous proinflammatory cytokines (e.g., interleukin-6 (IL-6) and IL-8), chemokines (e.g., monocyte chemoattractant proteins (MCPs) and macrophage inflammatory proteins (MIPs)), growth factors (e.g., transforming growth factor-β (TGFβ) and vascular endothelial growth factor (VEGF)), and proteases [25,26,27,28,29,30]. The secretion of those SASP factors can lead to accelerated aging in cancer survivors by triggering inflammation, promoting tumorigenesis and development of age-associated diseases, e.g., cognitive impairment [31] and CVD [32]. In this review, we focus on the mechanism of SASP promoting tumorigenesis.
SASP and Tumorigenesis
SASP factors can promote cancer cell proliferation, migration, invasiveness, angiogenesis, and epithelial-mesenchymal transition (EMT) [24]. Here, we briefly review the effect of SASP on each of these processes.
Evidence showed that SASP factors could promote cancer cell proliferation. For example, in the immunocompromised (nu/nu) mice model, tumors developed in seven out of 15 animals injected with senescent fibroblasts, while no tumors developed in eight animals injected with non-senescent fibroblasts [33]. One of the possible mechanisms is one of the SASP factors—IL-6—promotes tumor progression. IL-6 can promote proliferation by binding to the IL-6 receptor and subsequent activation of STAT3. STAT3 is widely considered as oncogene [34,35], and it can promote cancer progression through the transcription target genes [36].
SASP factors could promote cancer cell migration. For example, breast cancer cells showed the ability to migrate when induced with senescent fibroblasts [29]. This ability to migrate may be promoted by the secretion of SASP factors, e.g., IL-6 and IL-8, which can mediate the activation of STAT3.
SASP factors can promote EMT, an important step in cancer progression that allows the solid tumors to become more malignant, increasing their invasiveness and metastatic activity [37,38]. Coopé et al. incubated non-aggressive human breast cancer cell lines (T47D and ZR75.1) with conditioned media from senescent fibroblasts or pre-senescent fibroblasts induced by X-irradiation [29]. They found that fibroblast SASP induced a classic EMT in T47D and ZR75.1 cells [29]. Conditioned media from senescent cells markedly decreased overall and cell surface β-catenin and E-cadherin, and reduced cytokeratin expression, consistent with a mesenchymal transition. Furthermore, conditioned media from senescent cells downregulated the tight junction protein claudin-1, leaving the remaining protein localized primarily to the nucleus, a hallmark of an EMT [29]. IL-6 is likely to be one of the SASP factors that involved in the process of promoting EMT. Findings from the in vitro study showed IL-6 could induce EMT through the activation of STAT3 in human cervical carcinoma cells [39].
VEGF, one of the SASP factors, can promote tumor-driven angiogenesis, which is one of the hallmarks of cancer [40,41]. For example, in two groups of mice, a larger number and bigger sizes of vessels were observed in the group that was additionally injected with senescent mouse breast fibroblasts compared to the group that was injected with EpH4-v epithelial cells alone [40].
Although many commonly used cancer interventions induce senescence in tumor cells, SASP, a complex pro-inflammatory response of senescent cells, can lead to accelerated aging in cancer survivors by promoting various aspects of tumorigenesis and age-associated diseases.
2.2.2. Telomere Attrition
Telomeres are protein-bound DNA repeated structures at the ends of chromosomes that cap and stabilize the ends of chromosomes [42]. Telomeres shorten during each cycle of cellular division until the cell reaches its limited ability to divide. Telomerase, the enzyme, protects the telomere by maintaining telomere length in human cells [43,44]. As it decreases with age, telomere length has been proposed as a biomarker of aging [42,43,44].
Many cancer treatments impact telomere length, and some directly impair telomerase [45,46]. For example, chemotherapy can cause telomere shortening. It is not uncommon to see telomere length shortening among cancer survivors after receiving chemotherapy. Results from a study of 15 people with non-HL receiving conventional-dose chemotherapy showed that the mean telomere length decreased after chemotherapy (mean telomere length: 7.59 kb (before chemotherapy) vs. 7.07 kb (after chemotherapy), p = 0.03). The mean of their telomere length after chemotherapy was shorter compared to the mean of telomere length in their age-matched putatively cancer-free controls (mean telomere length: 8.71 kb (health controls) vs. 7.07 kb (people with cancer after chemotherapy), p < 0.01) [47]. In a study of 25 children with pediatric acute leukemia (n = 16) or solid tumor (n = 9), Engelhardt et al. found that the telomere length shortened in children after receiving chemotherapy [48]. This finding is in line with the laboratory studies of cancer cells line. In the study of BEL-7404 human hepatoma cell line, Zhang et al. found that the mean telomere lengths were decreased after the chemotherapy treatment (mean telomere length ranged from 3.1 to 4.1 kbp in the treated cells vs. 4.2 kbp of controls) [45].
In summary, many pieces of evidence indicated that cancer treatments could lead to a shorter telomere length, suggesting accelerated aging in cancer survivors.
2.2.3. Stem Cell Exhaustion
Stem cell exhaustion is the age-related reduction in activity of stem cells. Stem cells perform a wide range of functions, including the replacement of damaged or lost red blood cells and white blood cells [49,50]. In normal aging, the activity of stem cells decreases with age. For example, the National Marrow Donor Program (MNDP) study found that donor age was associated with lower overall and disease-free survival in bone marrow transplant recipients, suggesting that the activity of hematopoietic stem cells (HSCs) diminishes with age [51,52]. The stem cells may undergo an accelerated aging process. For example, after HCT, HSCs undergo replicative stress to allow hematopoietic reconstitution [53]. The replicative stress of HSCs occurring after HCT may lead to accelerated aging, as it can cause premature bone marrow failure, which is characterized by an inability to make enough blood cells, including red blood cells and white blood cells, in patients [54,55]. A single transplantation of HSCs was shown to effectively double the cellular aging that would occur normally [56].
2.2.4. DNA Damage
DNA damage, a critical factor in cancer development and progression, can also contribute to aging [57,58]. The DNA damage can be measured by γH2AX, which accumulates at the site of DNA damage and can be detected by immunocytochemistry [59,60]. Direct damage to DNA can be caused by the free radical intermediates generated by chemotherapeutic agents [61]. The alkylating agents, the oldest class of anticancer drugs, can lead to DNA double-strand breaks [57], while the DNA damage caused by alkylating agents is also associated with an increased risk of developing secondary leukemia in patients who treated with these agents [62,63]. For example, secondary leukemia has been reported in both children and older adults previously treated with alkylating agents [64,65]. Besides alkylating agents, cytotoxic drugs and ionizing radiation can also cause DNA damage [66].
2.2.5. Epigenetic Alterations
Many epigenetic alterations that are induced by cancer treatment drugs are likely to trigger the accelerated aging process. The well-known epigenetic modification, CpG island hypermethylation, is a component of drug-induced cytotoxicity. CpG island hypermethylation could cause inappropriate silencing of specific genes that may render the genome unstable and contribute to aging [67].
The examples of commonly used chemotherapy agents that can induce DNA hypermethylation at CpG islands in tumor cells include topoisomerase II inhibitors, doxorubicin, DNA cross-linking agents, and methotrexate [68]. For example, the alteration of 5-methylcytidine content was found in human lung epidermoid carcinoma cells after these cells were exposed to topoisomerase II inhibitors [68]. Some other drugs, including azacitidine and decitabine, hydralazine, and MG98, have also been reported as DNA hypermethylation agents. By inducing hypermethylation, all of these drugs may lead to accelerated aging in people with cancer [67].
In summary, these are the possible mechanisms that may explain the cancer treatment-induced accelerated aging in cancer survivors. At this time, most of the literature on accelerated aging from cellular senescence is based on data around the use of cytotoxic chemotherapy agents as well as radiation treatment. Further research is needed to clarify how targeted therapy and immunotherapy may impact cellular senescence.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13030427