Primary hyperoxalurias (PHs) are a group of inherited alterations of the hepatic glyoxylate metabolism. PHs classification based on gene mutations parallel a variety of enzymatic defects, and all involve the harmful accumulation of calcium oxalate crystals that produce systemic damage.
- hyperoxaluria
- oxalate
- inhibitor
- small molecule drug
- glycolate oxidase
- lactate dehydrogenase
- liver selective distribution
1. Primary Hyperoxalurias: Pathology and Current Treatment
Primary hyperoxaluria (PH) is a rare disease of liver metabolism that results in excess oxalate production and urine excretion (hyperoxaluria). Its estimated prevalence in clinical studies is around 1–3 per million population [1], although recent genomic investigations suggest significant underdiagnosis [2]. Indeed, genomic studies identify such number of mutant alleles in the general population that a prevalence of one in 58,000 individuals could be a good estimate [2]. This severe disease is caused by genetic changes that alter glyoxylate and hydroxyproline metabolism resulting in overproduction of oxalate by the liver [3]. Other situations in which elevated oxalate in the urine is due to excessive intake or absorption of oxalate or its precursor are known as secondary hyperoxalurias.
PH1 (OMIM #259900) is the most common and severe form of PH, due to mutations in the AGXT gene (coding for alanine: glyoxylate amino-transferase, AGT) [4] and accounts for about 80% of PH cases. Other hereditary hyperoxalurias include PH2 (OMIM #260000), caused by mutations in the GRHPR gene (coding for glyoxylate/hydroxypyruvate reductase, GRHPR) and PH3 (OMIM #613616), caused by mutations in the HOGA1 gene (coding 4-hydroxy-2-oxoglutarate aldolase 1, HOGA1). All three forms of PH are inherited as autosomal recessive diseases and the possibility exists that additional types of PH (non PH1-PH3) might be described in the future.
Loss of function mutations in any of these three genes result in a deficit to detoxify glyoxylate, which is then converted into oxalate by hepatic lactate dehydrogenase (LDH). Since humans have no enzyme capable of degrading oxalate, this dicarboxylic compound must be eliminated primarily by the kidneys, where it can complex with calcium to form crystals and calcium oxalate (CaOx) stones (Figure 1). Patients with primary hyperoxaluria experience high concentrations of oxalate in the urine from birth. PH1 patients typically yield oxalate excretion > 1 mmol/1.73 m2 per day (normal range < 0.5 mmol) [5]. The increased urinary excretion of oxalate results in urinary CaOx supersaturation, which leads to crystal aggregation, urolithiasis, and/or nephrocalcinosis. Nephrocalcinosis or recurrent urolithiasis can cause renal tubulointerstitial inflammation and fibrosis and, if persistent, end-stage kidney disease. In addition, urolithiasis complications such as infection and obstruction and the surgical interventions to treat urolithiasis, also contribute to renal damage in affected patients.
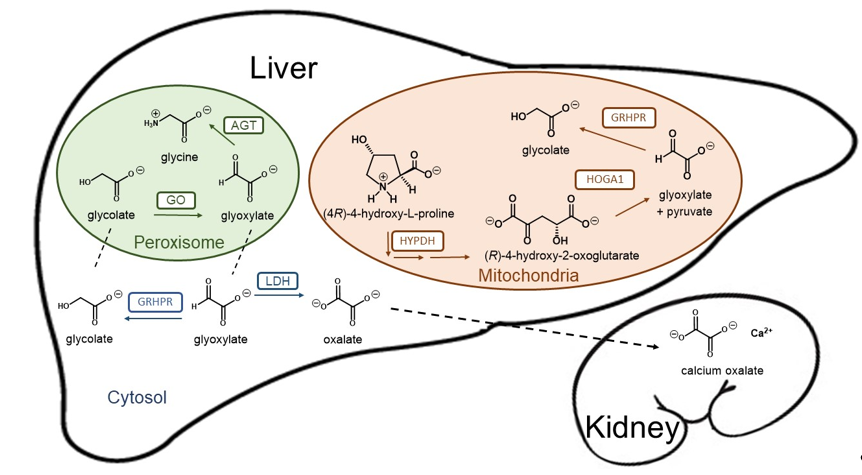
Figure 1. Schematic representation of the hepatic glyoxylate metabolism and calcium oxalate formation. Glyoxylate is a very reactive aldehyde produced in the intermediary metabolism of glycine, (4R)-4-hydroxy-L-proline and glycolate as the best known sources in humans. Lactate dehydrogenase (LDH) oxidizes cytosolic glyoxylate into oxalate, an end product of metabolism that can precipitate as tissue-damaging calcium oxalate. Two organelles play crucial roles in glyoxylate detoxification. Alanine-glyoxylate aminotransferase (AGT) plays a central role converting glyoxylate into glycine in the peroxisome, while mitochondrial and cytosolic glyoxylate can be reduced to glycolate by glycolate reductase / hydroxypyruvate reductase (GRHPR), preventing excessive oxidation to oxalate by LDH. Mitochondrial hydroxyproline metabolism via hydroxyproline dehydrogenase (HYPDH) results in the production of (R)-4-hydroxy-2-oxoglutarate that is normally split into glyoxylate and pyruvate by 4-hydroxy-2-oxoglutarate aldolase 1 (HOGA1).
The typical clinical presentation is either recurrent kidney stone episodes at a young age and/or chronic renal failure. In many instances, most frequently in PH1, oxalate nephropathy results in end stage renal disease (ESRD) (Figure 2). The consequences of declining kidney function can be particularly severe in these conditions, since the oxalate load can no longer be eliminated, and plasma oxalate levels rise to life-threatening levels [1]. In PH patients with kidney failure, dialysis cannot remove enough oxalate to keep up with daily production, resulting in oxalate deposition in skin, retina, heart, vessels, bones and other organs (systemic oxalosis), leading to severe morbidity and death.
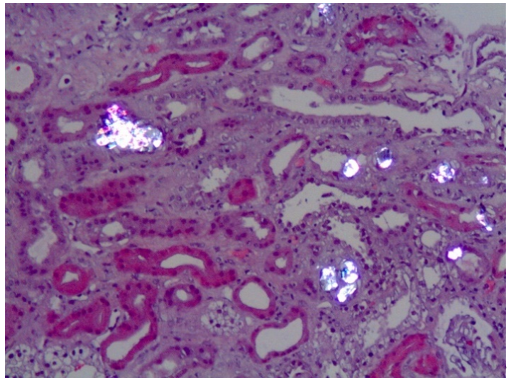
Figure 2. Calcium oxalate crystals (bright birefringent crystals) are shown in a kidney biopsy of a PH1 patient with chronic renal disease. Note the tubular damage and atrophy, interstitial inflammation and fibrosis secondary to calcium oxalate deposits.
Current strategies to reduce kidney damage try to reduce CaOx crystal formation through high urine volume and medications such as citrate and magnesium, and in some PH patients pharmacologic doses of pyridoxine can reduce oxalate production [6]. Very large daily fluid intake is required as well as medications taken multiple times daily, compromising quality of life. Further, while these treatments may decrease the effects of hyperoxaluria, in most PH patients they do not eliminate recurring stones or ESRD. The burden of frequent symptomatic kidney stones and their associated pain, hospitalizations and need for interventional procedures, and the burden of ESRD, intensive dialysis, systemic oxalate deposition, and transplantation is enormous. Kidney transplantation alone has a high failure rate due to oxalate injury. Thus, for the severe forms of PH combined liver and kidney transplantation is the only curative treatment to date but it has significant morbi-mortality and problems associated with donor organ shortage and life-long immunosuppressive treatment [6].
Substrate reduction therapy (SRT) targeting glycolate oxidase (GO) was proposed as a novel therapeutic approach [7][8] with no nocive effects [9][10][11]. Recently, the FDA and EMA agencies have approved the first pharmacological treatment for PH1, based on siRNA inhibition of GO (lumasiran), after it has revealed promising results, meeting its primary efficacy endpoint and all tested secondary endpoints.
2. Therapeutic Approaches in Development Against Primary Hyperoxalurias: Brief Overview
The arrival of lumasiran to the market means an important achievement towards the efficient pharmacological treatment of PH1 and constitutes the third example of siRNA drug approved for clinical use [12][13].
Many other therapeutic approaches are being currently developed against PHs in general and against PH1 specifically, being this one the most severe type of PH. Such approaches include gene/protein/cell therapies, small drug administration (chaperones and enzyme inhibitors) or the use of oxalate degrading bacteria or enzymes. These strategies are aimed at different targets, whether focused on producing a decrease of the plasmatic oxalate or on minimizing the renal damage subsequent to CaOx crystallization. An interesting classification of the therapeutic approaches attending to the organs or systems at which they are targeted can be found in the recent review by Kletzmayr et al. [14].
In summary, both, biotechnological therapeutic agents and small-molecule drugs, are under exploration for the treatment of PHs (Figure 3). Small molecule drugs constitute a classical approach and, in contrast to biopharmaceuticals, present the advantage of possible oral administration and, in general, lower production costs. Besides, more is known about the possible secondary outcomes upon chronic administration or administration to children [15]. Enzymatic inhibition using small drugs is a successful approach for the treatment of uncountable diseases. Being small drugs a promising therapeutic option, in this review we want to summarize the attention that is currently being paid to small-molecule drugs in the developing treatments for PHs.
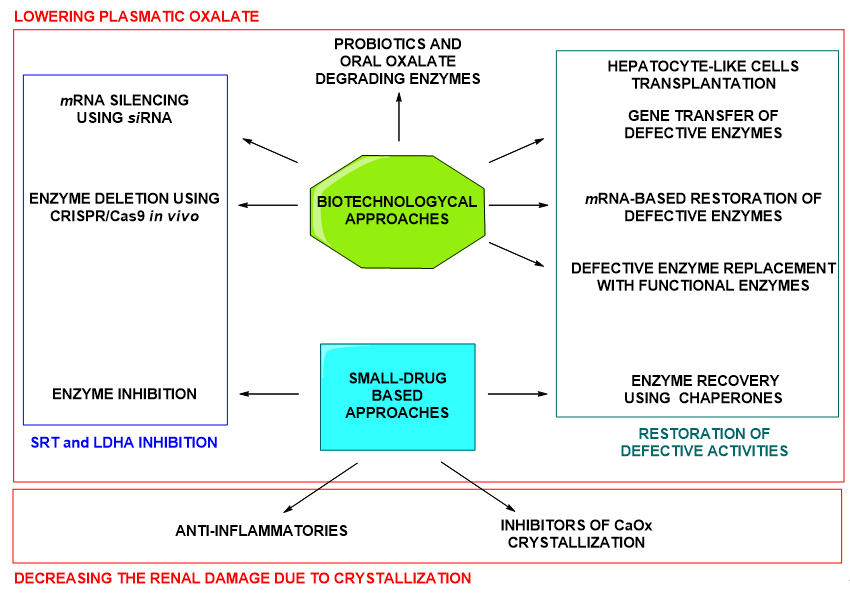
Figure 3. Summary of biopharmaceuticals and small drug approaches under development for the treatment of PHs.
2.1. Therapeutic Approaches Aimed at the Lowering of the Oxalate Plasmatic Concentration.
2.1.1. Recovery of Defective Activity
The aim of this approach is normalizing the glyoxylate altered metabolism by recovery of the defective activity.
Current research on cell therapy against PHs, pursues the transplantation of genetically modified autologous hepatocyte-like cells (HLCs), which are obtained from pluripotent stem cells of PH patients (PH-iPSCs). In preliminary investigations for PH1 treatment, these PH1-iPSCs have been successfully transformed ex vivo with a lentiviral vector encoding wild-type AGT, to obtain HLCs with significant AGT expression [16]. In this sense, CRISPR/Cas9 technology has become an essential tool to deliver Agxt gene in PH1-iPSCs [17][18]. Gene therapy is also being investigated as a therapeutic option for PH1 at this level [19]. Major progress has been made towards in vivo delivery of AGT. Effectively, injection of liver-directed vectors encoding AGT to AGT-deficient mice decreased the urinary oxalate and prevented oxalate crystalluria [20][21][22]. Protein therapy with wild-type AGT is another therapeutic option under research for PH1. In this direction, AGT variants with enhanced stability have been obtained leading the way to enzyme-replacement therapy in PH1 [23]. Besides, polypeptide-based AGT conjugates have successfully been internalized in the cellular model of PH1 CHO-GO, restoring AGT activity inside the peroxisomal compartment [24]. Moreover, against PH1, AGT mRNA constructs have been screened in vitro and in wild-type mice for the production of a functional AGT enzyme. Up to 40% reduction in urinary oxalate has been observed using this methodology, suggesting that mRNA encoding AGT led to increased expression and activity of the AGT enzyme in liver [25].
The recovery of the defective AGT activity is also being addressed by the use of small drugs. In this case, pharmacological chaperones have been observed to promote the correct folding of AGT and its correct localization in the subcellular compartments [26][27][28][29][30]. Pyridoxine is currently used in the treatment of PH1 as it is able to rescue the effect of misfolding mutations of AGT, though its effectivity is only associated to certain folding-defective AGT variants [31][32]. Pyridoxamine and pyridoxal can exert the same stabilizing effect [33], and so does aminooxoacetic acid [34][35].
2.1.2. Substrate Reduction Therapy (SRT)
Another important strategy is the inhibition of key enzymes involved in the production of glyoxylate. The aim is decreasing the hepatic concentration of glyoxylate, which is the substrate giving rise to oxalate formation in PHs. This SRT is being developed at the level of GO [7] and hydroxyproline dehydrogenase (HYPDH) inhibition [36][37]. While the first target gives rise to useful drugs against PH1, the second one is aimed against PH2 and PH3 [38][39]. GO catalyses the formation of glyoxylate from glycolate in the hepatic peroxisomes, where AGT is in charge of detoxifying glyoxylate in physiological conditions (Figure 1). In fact, silencing of GO mRNA with siRNA is the successful mechanism of the approved drug lumasiran [40][41]. Other results support the success of this strategy against PH1 in different studies in vivo (Agxt1-/- mice) [15][42][43][44]. Another biotechnological approach in exploration for GO silencing is the GO deletion using in vivo CRISPR/Cas9 technology [45][46]. However, GO inhibition is also being assessed with small-molecule drugs [47][48][49][50][51]. Recently, furylsalicylic derivatives have been reported to decrease oxalate production in Agxt1-/- mouse hepatocytes, being this phenotypic effect to some extent related to GO inhibition. However, a multiple-target mechanism was suggested for these molecules, possibly including lactate dehydrogenase A (LDHA) inhibition [49]. Other type of recently reported GO inhibitors (GOi’s) with nanomolar activity present triazole core structure [51].
More enzymatic inhibitors in development for SRT are HYPDH inhibitors [52]. HYPDH is the first enzyme in the hydroxyproline catabolism in liver and kidney towards glyoxylate formation, which is detoxified by the enzyme GRHPR in mitochondria and cytosol (Figure 1). Double Grhpr KO (PH2 model) and Prodh2 KO (HYPDH KO) mice showed no CaOx crystal deposition in kidneys when challenged with hydroxyproline in diet, supporting the utility of HYPDH as a target against PHs [39]. HYPDH inhibition by small molecules is under development and general structures of these molecules are available [52]. However, the structural information so far is not too specific and, for that reason, HYPDH inhibition is no further detailed in this review.
2.1.3. Lactate Dehydrogenase A (LDHA) Inhibition
Another possibility is the prevention of oxalate formation by direct inhibition/silencing of the enzyme in charge of its formation from the accumulated glyoxylate, the hepatic isozyme LDHA (Figure 1) [53][54]. Following this strategy, useful therapeutic agents might be found against the three types of PHs. In this sense, biotechnological agents (siRNA) as well as small inhibitors are being studied as potential drugs [55][56][57].
2.1.4. Regulating Oxalate Uptake/Secretion at Intestinal Level
Oral administration of probiotic bacteria or probiotics-derived factors are alternative therapeutic approaches under development against PHs [58][59][60][61][62]. These agents can degrade dietary oxalate, preventing its absorption at intestinal level. The consequence is a decrease of plasmatic and thus, urinary oxalate. Some species of Lactobacillus and Bifidobacterium are being studied [60][63][64]. Especially interesting is Oxalobacter formigenes as, besides its oxalate-degrading activity, it has been proved to be effective stimulating oxalate secretion by the intestinal epithelium helping its clearance from plasma, in PH1 mice [61][65][66] and human PH patients [67][68][69].
2.2. Therapeutic Approaches Aimed at the Minimization of the Renal Damage Provoked by CaOx Crystallization
The loss of renal functionality in PHs, is related to the inflammatory response produced by CaOx crystals deposition in the renal tubular cells, as it leads to chronic fibrogenesis. Thus, the anti-inflammatory therapy has been suggested as a therapeutic strategy in PHs as well as in other crystal accumulation pathologies [70][71]. It has been demonstrated that the inhibition of NLRP3, a component of the NALP3-inflammasome, using small molecules reduces the production of pro-inflammatory cytokines IL-1β and IL-18 and this effectively prevents kidney fibrosis and attenuates renal inflammation in mouse models, without immunosuppressive side effects [72]. The same way, small antagonists of IL-1β [73] and TNF-α [74] receptors, have been seen to protect from CaOx nephropathy in mice.
Inhibition of CaOx crystallization is another strategy under research aimed at the elimination of the renal damage caused by crystal deposition [75][76][77]. In this sense, important advances have been made recently, with the discovery of a new powerful class of CaOx inhibitors based in multivalent inositol phosphate molecules that can inhibit the crystallization process as well as the CaOx-cell interactions that lead to kidney damage [78].
This entry is adapted from the peer-reviewed paper 10.3390/jpm11020074
References
- Cochat, P.; Rumsby, G. Primary Hyperoxaluria. Engl. J. Med. 2013, 369, 649–658, doi:10.1056/nejmra1301564.
- Hopp, K.; Cogal, A.G.; Bergstralh, E.J.; Seide, B.M.; Olson, J.B.; Meek, A.M.; Lieske, J.C.; Milliner, D.S.; Harris, P.C. Phenotype-Genotype Correlations and Estimated Carrier Frequencies of Primary Hyperoxaluria. Am. Soc. Nephrol. 2015, 26, 2559–2570, doi:10.1681/asn.2014070698.
- Salido, E.; Pey, A.L.; Rodriguez, R.; Lorenzo, V. Primary hyperoxalurias: Disorders of glyoxylate detoxification. Biophys. Acta BBA Mol. Basis Dis. 2012, 1822, 1453–1464, doi:10.1016/j.bbadis.2012.03.004.
- Milliner, D.S.; Harris, P.C.; Cogal, A.G.; Lieske, J.C. Primary Hyperoxaluria Type 1. In Gene Reviews, 2nd ed; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace SE, Bean LJH, Stephens K, Amemiya A., Eds.; University of Washington: Seattle, DC, USA, 2017; Available online: https://www.ncbi.nlm.nih.gov/books/NBK1283/ (accessed on 11 December 2020).
- Hoppe, B.; Beck, B.B.; Milliner, D.S. The primary hyperoxalurias. Kidney Int. 2009, 75, 1264–1271, doi:10.1038/ki.2009.32.
- Cochat, P.; Hulton, S.-A.; Acquaviva, C.; Danpure, C.J.; Daudon, M.; De Marchi, M.; Fargue, S.; Groothoff, J.; Harambat, J.; Hoppe, B.; et al. Primary hyperoxaluria Type 1: Indications for screening and guidance for diagnosis and treatment. Dial. Transplant. 2012, 27, 1729–1736, doi:10.1093/ndt/gfs078.
- Martin-Higueras, C.; Luis-Lima, S.; Salido, E. Glycolate Oxidase Is a Safe and Efficient Target for Substrate Reduction Therapy in a Mouse Model of Primary Hyperoxaluria Type I. Ther. 2016, 24, 719–725, doi:10.1038/mt.2015.224.
- Dutta, C.; Avitahl-Curtis, N.; Pursell, N.; Cohen, M.L.; Holmes, B.; Diwanji, R.; Zhou, W.; Apponi, L.; Koser, M.; Ying, B.; et al. Inhibition of Glycolate Oxidase With Dicer-substrate siRNA Reduces Calcium Oxalate Deposition in a Mouse Model of Primary Hyperoxaluria Type 1. Ther. 2016, 24, 770–778, doi:10.1038/mt.2016.4.
- Frishberg, Y.; Zeharia, A.; Lyakhovetsky, R.; Bargal, R.; Belostotsky, R. Mutations inHAO1encoding glycolate oxidase cause isolated glycolic aciduria. Med. Genet. 2014, 51, 526–529, doi:10.1136/jmedgenet-2014-102529.
- McGregor, T.L.; Hunt, K.A.; Nioi, P.; Mason, D.; Ticau, S.; Pelosi, M.; Loken, P.R.; Finer, S.; Griffiths, C.J.; MacArthur, D.G.; et al. Deep Phenotyping of a Healthy Human HAO1 Knockout Informs Therapeutic Development for Primary Hyperoxaluria Type 1. bioRxiv 2019, 524256, doi:10.1101/524256.
- McGregor, T.L.; Hunt, K.A.; Yee, E.; Mason, D.; Nioi, P.; Ticau, S.; Pelosi, M.; Loken, P.R.; Finer, S.; Lawlor, D.A.; et al. Characterising a healthy adult with a rare HAO1 knockout to support a therapeutic strategy for primary hyperoxaluria. eLife 2020, 9, doi:10.7554/elife.54363.
- Garber, K. Alnylam launches era of RNAi drugs. Biotechnol. 2018, 36, 777–778, doi:10.1038/nbt0918-777.
- Second RNAi drug approved. Biotechnol. 2020, 38, 385, doi:10.1038/s41587-020-0494-3.
- Kletzmayr, A.; Ivarsson, M.E.; Leroux, J.-C. Investigational Therapies for Primary Hyperoxaluria. Bioconjugate Chem. 2020, 31, 1696–1707, doi:10.1021/acs.bioconjchem.0c00268.
- Milliner, D.S. siRNA Therapeutics for Primary Hyperoxaluria: A Beginning. Ther. 2016, 24, 666–667, doi:10.1038/mt.2016.50.
- Estève, J.; Blouin, J.-M.; Lalanne, M.; Azzi-Martin, L.; Dubus, P.; Bidet, A.; Harambat, J.; Llanas, B.; Moranvillier, I.; Bedel, A.; et al. Generation of induced pluripotent stem cells-derived hepatocyte-like cells for ex vivo gene therapy of primary hyperoxaluria type 1. Stem Cell Res. 2019, 38, 101467, doi:10.1016/j.scr.2019.101467.
- Estève, J.; Blouin, J.-M.; Lalanne, M.; Azzi-Martin, L.; Dubus, P.; Bidet, A.; Harambat, J.; Llanas, B.; Moranvillier, I.; Bedel, A.; et al. Targeted gene therapy in human-induced pluripotent stem cells from a patient with primary hyperoxaluria type 1 using CRISPR/Cas9 technology. Biophys. Res. Commun. 2019, 517, 677–683, doi:10.1016/j.bbrc.2019.07.109.
- Zheng, R.; Li, Y.; Wang, L.; Fang, X.; Zhang, J.; He, L.; Yang, L.; Li, D.; Geng, H. CRISPR/Cas9–mediated metabolic pathway reprogramming in a novel humanized rat model ameliorates primary hyperoxaluria type 1. Kidney Int. 2020, 98, 947–957, doi:10.1016/j.kint.2020.04.049.
- Koul, S.; Johnson, T.; Pramanik, S.; Koul, H.K. Cellular transfection to deliver alanine-glyoxylate aminotransferase to hepatocytes: A rational gene therapy for primary hyperoxaluria-1 (PH-1). J. Nephrol. 2005, 25, 176–182, doi:10.1159/000085410.
- Salido, E.C.; Li, X.M.; Lu, Y.; Wang, X.; Santana, A.; Roy-Chowdhury, J.; Torres, A.; Shapiro, L.J. Alanine-glyoxylate aminotransferase-deficient mice, a model for primary hyperoxaluria that responds to adenoviral gene transfer. Natl. Acad. Sci. USA 2006, 103, 18249–18254.
- Salido, E.; Rodriguez-Pena, M.; Santana, A.; Beattie, S.G.; Petry, H.; Torres, A. Phenotypic Correction of a Mouse Model for Primary Hyperoxaluria With Adeno-associated Virus Gene Transfer. Ther. 2011, 19, 870–875, doi:10.1038/mt.2010.270.
- Castello, R.; Borzone, R.; D’Aria, S.; Annunziata, P.; Piccolo, P.; Brunetti-Pierri, N. Helper-dependent adenoviral vectors for liver-directed gene therapy of primary hyperoxaluria type 1. Gene Ther. 2016, 23, 129–134, doi:10.1038/gt.2015.107.
- Mesa-Torres, N.; Yunta, C.; Fabelo‑Rosa, I.; González-Rubio, J.M.; Sanchez-Ruiz, J.M.; Salido, E.; Albert, A.; Pey, A.L. The consensus-based approach for gene/enzyme replacement therapies and crystallization strategies: The case of human alanine–glyoxylate aminotransferase. J. 2014, 462, 453–463, doi:10.1042/bj20140250.
- Roncador, A.; Oppici, E.; Talelli, M.; Pariente, A.N.; Donini, M.; Dusi, S.; Voltattorni, C.B.; Vicent, M.J.; Cellini, B. Use of polymer conjugates for the intraperoxisomal delivery of engineered human alanine:glyoxylate aminotransferase as a protein therapy for primary hyperoxaluria type I. Nanotechnol. Biol. Med. 2017, 13, 897–907, doi:10.1016/j.nano.2016.12.011.
- Kukreja, A.; Lasaro, M.; Cobaugh, C.; Forbes, C.; Tang, J.-P.; Gao, X.; Martin-Higueras, C.; Pey, A.L.; Salido, E.; Sobolov, S.; et al. Systemic Alanine Glyoxylate Aminotransferase mRNA Improves Glyoxylate Metabolism in a Mouse Model of Primary Hyperoxaluria Type 1. Nucleic Acid Ther. 2019, 29, 104–113, doi:10.1089/nat.2018.0740.
- Miyata, N.; Steffen, J.; Johnson, M.E.; Fargue, S.; Danpure, C.J.; Koehler, C.M. Pharmacologic rescue of an enzyme-trafficking defect in primary hyperoxaluria 1. Natl. Acad. Sci. USA 2014, 111, 14406–14411.
- Oppici, E.; Montioli, R.; Dindo, M.; Cellini, B. Natural and Unnatural Compounds Rescue Folding Defects of Human Alanine: Glyoxylate Aminotransferase Leading to Primary Hyperoxaluria Type I. Drug Targets 2016, 17, 1482–1491, doi:10.2174/1389450117666160302095254.
- Hou, S.; Madoux, F.; Scampavia, L.; Janovick, J.A.; Conn, P.M.; Spicer, T.P. Drug Library Screening for the Identification of Ionophores That Correct the Mistrafficking Disorder Associated with Oxalosis Kidney Disease. SLAS Discov. Adv. Life Sci. R D 2017, 22, 887–896, doi:10.1177/2472555217689992.
- Yang, H.; Male, M.; Li, Y.; Wang, N.; Zhao, C.; Jin, S.; Jin, S.; Chen, Z.; Zhang, X.; Xu, H. Efficacy of Hydroxy-L-proline (HYP) analogs in the treatment of primary hyperoxaluria in Drosophila Melanogaster. BMC Nephrol. 2018, 19, 167, doi:10.1186/s12882-018-0980-8.
- Dindo, M.; Grottelli, S.; Annunziato, G.; Giardina, G.; Pieroni, M.; Pampalone, G.; Faccini, A.; Cutruzzolà, F.; Laurino, P.; Costantino, G.; et al. Cycloserine enantiomers are reversible inhibitors of human alanine:glyoxylate aminotransferase: Implications for Primary Hyperoxaluria type 1. J. 2019, 476, 3751–3768, doi:10.1042/bcj20190507.
- Monico, C.G.; Rossetti, S.; Olson, J.B.; Milliner, D.S. Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int. 2005, 67, 1704–1709, doi:10.1111/j.1523-1755.2005.00267.x.
- Dindo, M.; Oppici, E.; Dell’Orco, D.; Montone, R.; Cellini, B. Correlation between the molecular effects of mutations at the dimer interface of alanine–glyoxylate aminotransferase leading to primary hyperoxaluria type I and the cellular response to vitamin B6. Inherit. Metab. Dis. 2018, 41, 263–275, doi:10.1007/s10545-017-0105-8.
- Oppici, E.; Fargue, S.; Reid, E.S.; Mills, P.; Clayton, P.T.; Danpure, C.J.; Cellini, B. Pyridoxamine and pyridoxal are more effective than pyridoxine in rescuing folding-defective variants of human alanine:glyoxylate aminotransferase causing primary hyperoxaluria type I. Mol. Genet. 2015, 24, 5500–5511, doi:10.1093/hmg/ddv276.
- Oppici, E.; Montioli, R.; Dindo, M.; Maccari, L.; Porcari, V.; Lorenzetto, A.; Chellini, S.; Voltattorni, C.B.; Cellini, B. The Chaperoning Activity of Amino-oxyacetic Acid on Folding-Defective Variants of Human Alanine:Glyoxylate Aminotransferase Causing Primary Hyperoxaluria Type I. ACS Chem. Biol. 2015, 10, 2227–2236, doi:10.1021/acschembio.5b00480.
- Horváth, V.P.; Wanders, R.J. Aminooxy acetic acid: A selective inhibitor of alanine:glyoxylate aminotransferase and its use in the diagnosis of primary hyperoxaluria type I. Chim. Acta 1995, 243, 105–114, doi:10.1016/0009-8981(95)06149-5.
- Summitt, C.B.; Johnson, L.C.; Jönsson, T.J.; Parsonage, D.; Holmes, R.P.; Lowther, W.T. Proline dehydrogenase 2 (PRODH2) is a hydroxyproline dehydrogenase (HYPDH) and molecular target for treating primary hyperoxaluria. J. 2015, 466, 273–281, doi:10.1042/bj20141159.
- Lowther, T.; Holmes, R. Combinations for the Treatment of Kidney Stones. U.S. Patent WO2017100268 (A1), 15-June-2017.
- Fargue, S.; Milliner, D.S.; Knight, J.; Olson, J.B.; Lowther, W.T.; Holmes, R.P. Hydroxyproline Metabolism and Oxalate Synthesis in Primary Hyperoxaluria. Am. Soc. Nephrol. 2018, 29, 1615–1623, doi:10.1681/asn.2017040390.
- Buchalski, B.; Wood, K.D.; Challa, A.; Fargue, S.; Holmes, R.P.; Lowther, W.T.; Knight, J. The effects of the inactivation of Hydroxyproline dehydrogenase on urinary oxalate and glycolate excretion in mouse models of primary hyperoxaluria. Biophys. Acta BBA Mol. Basis Dis. 2020, 1866, 165633, doi:10.1016/j.bbadis.2019.165633.
- Querbes, W.; Fitzgerald, K.; Bettencourt, B.; Liebow, A.; Erbe, D. Compositions and Methods for Inhibition of Hao1 (Hy-droxyacid Oxidase 1 (Glycolate Oxidase)) Gene Expression. U.S. Patent WO2016057893 (A1), 14-April-2016.
- Erbe, D. Methods for Inhibition of Hao1 (Hydroxyacid Oxidase 1 (Glycolate Oxidase) Gene Expression. U.S. Patent WO2019014491 (A1), 17-January-2019.
- Brown, B.; Dudek, H. Methods and Compositions for the Specific Inhibition of Glycolate Oxidase (Hao1) by Dou-ble-Stranded Rna. U.S. Patent WO2015100436 (A1), 02-July-2015.
- Li, X.; Knight, J.; Fargue, S.; Buchalski, B.; Guan, Z.; Inscho, E.W.; Liebow, A.; Fitzgerald, K.; Querbes, W.; Lowther, W.T.; et al. Metabolism of 13C5-hydroxyproline in mouse models of Primary Hyperoxaluria and its inhibition by RNAi therapeutics targeting liver glycolate oxidase and hydroxyproline dehydrogenase. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 233–239, doi:10.1016/j.bbadis.2015.12.001.
- Liebow, A.; Li, X.; Racie, T.; Hettinger, J.; Bettencourt, B.R.; Najafian, N.; Haslett, P.; Fitzgerald, K.; Holmes, R.P.; Erbe, D.; et al. An Investigational RNAi Therapeutic Targeting Glycolate Oxidase Reduces Oxalate Production in Models of Primary Hyperoxaluria. Am. Soc. Nephrol. 2016, 28, 494–503, doi:10.1681/asn.2016030338.
- Zabaleta, N.; Barberia, M.; Martin-Higueras, C.; Zapata-Linares, N.; Betancor, I.; Rodriguez, S.; Martinez-Turrillas, R.; Torella, L.; Vales, A.; Olagüe, C.; et al. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Commun. 2018, 9, 1–9, doi:10.1038/s41467-018-07827-1.
- Davey, R.; Jantz, D.; Smith, J.J.; Owens, G. Genetic Modification of the Hydroxyacid Oxidase 1 Gene for Treatment of Primary Hyperoxaluria. U.S. Patent WO2020132659 (A1), 25-June-2020.
- Wang, M.; Xu, M.; Long, Y.; Fargue, S.; Southall, N.; Hu, X.; McKew, J.C.; Danpure, C.J.; Zheng, W. High throughput cell-based assay for identification of glycolate oxidase inhibitors as a potential treatment for Primary Hyperoxaluria Type 1. Rep. 2016, 6, srep34060, doi:10.1038/srep34060.
- Lowther, W.T.; Holmes, R.P. Glycolate Oxidase Inhibitors and Methods of Use for the Treatment of Kidney Stones. U.S. Patent WO2017100266 (A1), 15-June-2017.
- Moya-Garzón, M.D.; Martin-Higueras, C.; Peñalver, P.; Romera, M.; Fernandes, M.X.; Franco-Montalban, F.; Gómez-Vidal, J.A.; Salido, E.; Díaz-Gavilán, M. Salicylic Acid Derivatives Inhibit Oxalate Production in Mouse Hepatocytes with Primary Hyperoxaluria Type 1. Med. Chem. 2018, 61, 7144–7167, doi:10.1021/acs.jmedchem.8b00399.
- Wang, B.; Chao, Q. Glycolate Oxidase Inhibitors for the Treatment of Disease. U.S. Patent WO2019133770 (A2), 04-July-2019.
- Maag, H.; Fernandes, M.X.; Zamboni, R.; Akbariromani, E.; Beaulieu, M.-A.; Leblanc, Y.; Thakur, P. Triazole Glycolate Oxidase Inhibitors. U.S. Patent WO2020010309 (A1), 09-January-2020.
- Lowther, W.T.; Holmes, R.P. Hypdh Inhibitors and Methods of Use for the Treatment of Kidney Stones. U.S. Patent WO2016123012 (A1), 04-August-2016.
- Stevens, J.S.; Al-Awqati, Q. Lactate dehydrogenase 5: Identification of a druggable target to reduce oxaluria. Clin. Investig. 2019, 129, 2201–2204, doi:10.1172/jci128709.
- Letavernier, E.; Daudon, M. Stiripentol identifies a therapeutic target to reduce oxaluria. Opin. Nephrol. Hypertens. 2020, 29, 394–399, doi:10.1097/mnh.0000000000000621.
- Lai, C.; Pursell, N.; Gierut, J.; Saxena, U.; Zhou, W.; Dills, M.; Diwanji, R.; Dutta, C.; Koser, M.; Nazef, N.; et al. Specific Inhibition of Hepatic Lactate Dehydrogenase Reduces Oxalate Production in Mouse Models of Primary Hyperoxaluria. Ther. 2018, 26, 1983–1995, doi:10.1016/j.ymthe.2018.05.016.
- Wood, K.D.; Holmes, R.P.; Erbe, D.; Liebow, A.; Fargue, S.; Knight, J. Reduction in urinary oxalate excretion in mouse models of Primary Hyperoxaluria by RNA interference inhibition of liver lactate dehydrogenase activity. Biophys. Acta BBA Mol. Basis Dis. 2019, 1865, 2203–2209, doi:10.1016/j.bbadis.2019.04.017.
- Le Dudal, M.; Huguet, L.; Perez, J.; Vandermeersch, S.; Bouderlique, E.; Tang, E.; Martori, C.; Chemaly, N.; Nabbout, R.; Haymann, J.-P.; et al. Stiripentol protects against calcium oxalate nephrolithiasis and ethylene glycol poisoning. Clin. Investig. 2019, 129, 2571–2577, doi:10.1172/jci99822.
- Grujic, D.; Salido, E.; Shenoy, B.C.; Langman, C.B.; McGrath, M.E.; Patel, R.J.; Rashid, A.; Mandapati, S.; Jung, C.W.; Margolin, A.L. Hyperoxaluria Is Reduced and Nephrocalcinosis Prevented with an Oxalate-Degrading Enzyme in Mice with Hyperoxaluria. J. Nephrol. 2009, 29, 86–93, doi:10.1159/000151395.
- Abratt, V.R.; Reid, S.J. Chapter 3—Oxalate-Degrading Bacteria of the Human Gut as Probiotics in the Management of Kidney Stone Disease. In Advances in Applied Microbiology; Laskin, A.I., Sariaslani, S., Gadd, G.M., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 72, pp. 63–87.
- Selle, K.; Klaenhammer, T.R. Genomic and phenotypic evidence for probiotic influences ofLactobacillus gasserion human health. FEMS Microbiol. Rev. 2013, 37, 915–935, doi:10.1111/1574-6976.12021.
- Whittamore, J.M.; Hatch, M. The role of intestinal oxalate transport in hyperoxaluria and the formation of kidney stones in animals and man. Urolithiasis 2017, 45, 89–108, doi:10.1007/s00240-016-0952-z.
- Russ, Z.; Whitaker, W.; Deloache, W.; Stanley, S.E. Methods and Compositions for Treating Hyperoxaluria. U.S. Patent WO2020123483 (A1), 18-June-2020.
- Kostovcikova, K.; Whittamore, J.M.; Hatch, M. Bifidobacterium animalis subsp. lactis decreases urinary oxalate excretion in a mouse model of primary hyperoxaluria. Urolithiasis 2015, 43, 107–117, doi:10.1007/s00240-014-0728-2.
- Chamberlain, C.A.; Hatch, M.; Garrett, T.J. Metabolomic profiling of oxalate-degrading probiotic Lactobacillus acidophilus and Lactobacillus gasseri. PLoS ONE 2019, 14, e0222393, doi:10.1371/journal.pone.0222393.
- Hatch, M.; Gjymishka, A.; Salido, E.C.; Allison, M.J.; Freel, R.W. Enteric oxalate elimination is induced and oxalate is normalized in a mouse model of primary hyperoxaluria following intestinal colonization withOxalobacter. J. Physiol. Liver Physiol. 2011, 300, G461–G469, doi:10.1152/ajpgi.00434.2010.
- Arvans, D.; Jung, Y.-C.; Antonopoulos, D.; Koval, J.; Granja, I.; Bashir, M.; Karrar, E.; Roy-Chowdhury, J.; Musch, M.; Asplin, J.; et al. Oxalobacter formigenes–Derived Bioactive Factors Stimulate Oxalate Transport by Intestinal Epithelial Cells. Am. Soc. Nephrol. 2016, 28, 876–887, doi:10.1681/asn.2016020132.
- Pape, L.; Ahlenstiel-Grunow, T.; Birtel, J.; Krohne, T.U.; Hoppe, B. Oxalobacter formigenes treatment combined with intensive dialysis lowers plasma oxalate and halts disease progression in a patient with severe infantile oxalosis. Nephrol. 2020, 35, 1121–1124, doi:10.1007/s00467-019-04463-9.
- Tavasoli, S.; Alebouyeh, M.; Naji, M.; Majd, G.S.; Nashtaei, M.S.; Broumandnia, N.; Basiri, A. Association of intestinal oxalate-degrading bacteria with recurrent calcium kidney stone formation and hyperoxaluria: A case-control study. BJU Int. 2020, 125, 133–143, doi:10.1111/bju.14840.
- Milliner, D.S.; Hoppe, B.; Groothoff, J. A randomised Phase II/III study to evaluate the efficacy and safety of orally administered Oxalobacter formigenes to treat primary hyperoxaluria. Urolithiasis 2018, 46, 313–323, doi:10.1007/s00240-017-0998-6.
- Martin-Higueras, C.; Ludwig-Portugall, I.; Hoppe, B.; Kurts, C. Targeting kidney inflammation as a new therapy for primary hyperoxaluria? Dial. Transplant. 2018, 34, 908–914, doi:10.1093/ndt/gfy239.
- Chen, Z.; Yuan, P.; Sun, X.; Tang, K.; Liu, H.; Han, S.; Ye, T.; Liu, X.; Yang, X.; Zeng, J.; et al. Pioglitazone decreased renal calcium oxalate crystal formation by suppressing M1 macrophage polarization via the PPAR-γ-miR-23 axis. J. Physiol. Physiol. 2019, 317, F137–F151, doi:10.1152/ajprenal.00047.2019.
- Ludwig-Portugall, I.; Bartok, E.; Dhana, E.; Evers, B.D.G.; Primiano, M.J.; Hall, J.P.; Franklin, B.S.; Knolle, P.; Hornung, V.; Hartmann, G.; et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016, 90, 525–539, doi:10.1016/j.kint.2016.03.035.
- Mulay, S.R.; Kulkarni, O.P.; Rupanagudi, K.V.; Migliorini, A.; Darisipudi, M.N.; Vilaysane, A.; Muruve, D.; Shi, Y.; Munro, F.; Liapis, H.; et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. Clin. Investig. 2012, 123, 236–246, doi:10.1172/jci63679.
- Mulay, S.R.; Eberhard, J.N.; Desai, J.; Marschner, J.A.; Kumar, S.V.; Weidenbusch, M.; Grigorescu, M.; Lech, M.; Eltrich, N.; Müller, L.; et al. Hyperoxaluria Requires TNF Receptors to Initiate Crystal Adhesion and Kidney Stone Disease. Am. Soc. Nephrol. 2016, 28, 761–768, doi:10.1681/asn.2016040486.
- Norman, R.W.; Scurr, D.S.; Robertson, W.G.; Peacock, M. Sodium pentosan polysulphate as a polyanionic inhibitor of calcium oxalate crystallization in vitro and in vivo. Sci. 1985, 68, 369–371, doi:10.1042/cs0680369.
- Alamani, B.G.; Rimer, J.D. Molecular modifiers of kidney stones. Opin. Nephrol. Hypertens. 2017, 26, 256–265, doi:10.1097/mnh.0000000000000330.
- Steiger, S.; Grill, J.F.; Ma, Q.; Bäuerle, T.; Jordan, J.; Smolle, M.; Böhland, C.; Lech, M.; Anders, H.-J. Anti-Transforming Growth Factor β IgG Elicits a Dual Effect on Calcium Oxalate Crystallization and Progressive Nephrocalcinosis-Related Chronic Kidney Disease. Immunol. 2018, 9, doi:10.3389/fimmu.2018.00619.
- Kletzmayr, A.; Mulay, S.R.; Motrapu, M.; Luo, Z.; Anders, H.; Ivarsson, M.E.; Leroux, J.-C. Inhibitors of Calcium Oxalate Crystallization for the Treatment of Oxalate Nephropathies. Sci. 2020, 7, 1903337, doi:10.1002/advs.201903337.