Extracellular signal-regulated kinase (ERK5) is an essential regulator of cancer progression, tumor relapse, and poor patient survival. Epithelial to mesenchymal transition (EMT) is a complex oncogenic process, which drives cell invasion, stemness, and metastases. Activators of ERK5, including mitogen-activated protein kinase 5 (MEK5), tumor necrosis factor α (TNF-α), and transforming growth factor-β (TGF-β), are known to induce EMT and metastases in breast, lung, colorectal, and other cancers. Several downstream targets of the ERK5 pathway, such as myocyte-specific enhancer factor 2c (MEF2C), activator protein-1 (AP-1), focal adhesion kinase (FAK), and c-Myc, play a critical role in the regulation of EMT transcription factors SNAIL, SLUG, and β-catenin. Moreover, ERK5 activation increases the release of extracellular matrix metalloproteinases (MMPs), facilitating breakdown of the extracellular matrix (ECM) and local tumor invasion. Targeting the ERK5 signaling pathway using small molecule inhibitors, microRNAs, and knockdown approaches decreases EMT, cell invasion, and metastases via several mechanisms.
- ERK5
- EMT
- cancer metastases
1. Introduction
The epithelial to mesenchymal transition (EMT), one of the first steps in cancer metastases, is a continuum of morphologic transitions from a cobblestone-like epithelial state to a spindle-like mesenchymal state. The complex process of metastases involves EMT, intravasation in the blood vessels, survival in blood stream, extravasation at the secondary site, mesenchymal to epithelial transition (MET), and secondary tumor growth. Cancer metastases require high cellular plasticity and adaptability to survive in diverse physiological environments. It is important to identify targeted therapy to target different stages of metastases. Upregulation of EMT transcription factors via growth factors, epigenetic plasticity, and downregulation of tumor suppressor microRNAs (miRs) are a few mechanisms that drive EMT in cancer [1,2].
Epithelial and mesenchymal cells are characterized by differences in molecular markers expressed by these cells. E-cadherin and cytokeratins are markers of epithelial phenotype, whereas mesenchymal cells express N-cadherin, SNAIL, SLUG, and Vimentin among others. These markers can be co-expressed during cancer progression, leading to an intermediate epithelial/mesenchymal state, which is associated with poor prognosis [3]. EMT is regulated in several developmental and fibrotic processes during embryogenesis (Type-I EMT) and wound healing (Type-II EMT), respectively [4]. These processes are regulated by different context-dependent signals, for example, transforming growth factor (TGF)-β [5]. However, the discussion of these studies is beyond the scope of the current review and the focus of the current review is to discuss EMT in cancer (Type-III EMT). Upregulation of mesenchymal markers can occur due to mutations in components of the KRAS, Wnt, or EGFR signaling pathways. The suppression of E-cadherin is a central event during EMT. Epidermal growth factor (EGF) can mediate transcriptional activation of SNAIL via p21-activated kinase (PAK) [6]. TGF-β and RAS can co-operate to induce EMT in breast cancer [7]. Wnt/β-catenin, Jagged (JAG), and sonic hedgehog (SHH) signaling pathways are some examples of ligand-mediated signaling events, which can activate EMT in cancer via activation of downstream targets T-cell factor/lymphoid enhancer factor (TCF/LEF), NOTCH, or Gli-1 [8]. Several mechanisms that regulate EMT in cancer are reviewed in Figure 1.
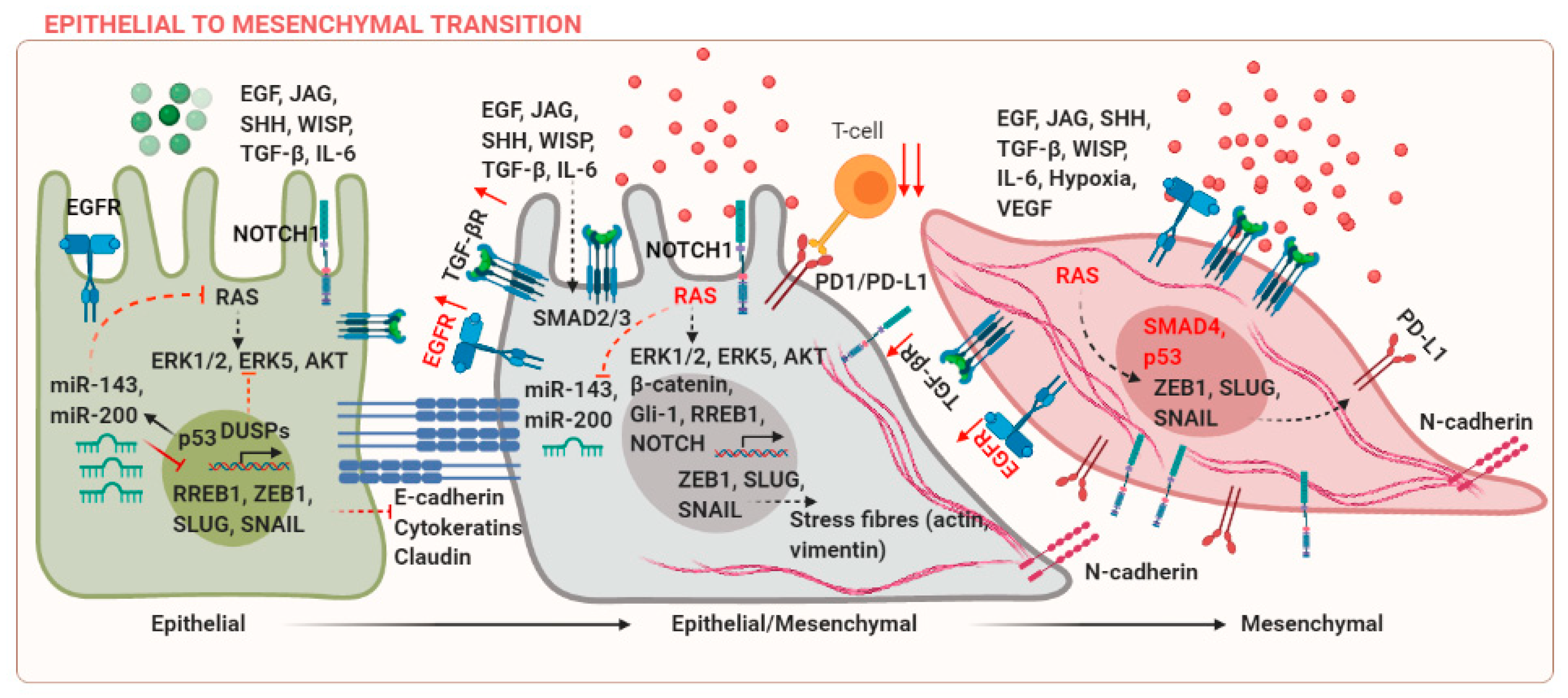
Tumor heterogeneity in terms of difference in driver mutations within the same cancer subtype and complexity of the tumor microenvironment has made the application of oncology therapeutics extremely challenging. Extracellular signaling factors and epigenetic effectors cooperate to initiate the EMT program and ultimately lead to metastases. One of the cellular adaptations during EMT involves increased capabilities of cancer cells to preferentially interact with the extracellular matrix, rather than the adjacent epithelial and stromal cells. Integrins are known to activate Src and focal adhesion kinases (FAK), which results in an increase in secretion of matrix metalloproteinases, loss of E-cadherin, and disruption of the adherens junctions (AJs) [9].
Tumor-associated chronic inflammation could initiate EMT via crosstalk between the inflammatory and tumor cells [10]. The tumor is infiltrated by diverse inflammatory and immune mediators. For example, 50% of the tumor is infiltrated with inflammatory macrophages. These infiltrated activated macrophages and proinflammatory T-cells can release cytokines, including transforming growth factor-β (TGF-β), tumor necrosis factor α (TNF-α), and interleukin-6 (IL-6), which are potent EMT inducers [11]. Expression of immune checkpoint proteins such as PD-L1 mediates the escape of cancer cells from natural killer (NK) cell-mediated cytotoxicity. In the given feedforward mechanism, EMT increases in PD-L1 expression via the microRNA-200-ZEB1 axis, which results in immune suppression and metastases [12].
Another feature that the cancer cells adopt as they transition to a mesenchymal state and enter the blood circulation for metastases is their ability to activate and bind platelets. At this stage, the cancer cells are termed as circulating tumor cells. Many studies argue that the cells are in an intermediate epithelial/mesenchymal state at this stage. There are coagulation-dependent mechanisms modulated via fibrin, which help the cancer cells to bind platelets and gain protection against the loss of anchorage-triggering anoikis, immune attack, and shear stress [13]. Drug resistant stem cells at the primary and metastatic sites are also known to possess EMT characteristics [14].
MicroRNA (miRNA) dysregulation is an important event during tumor progression. MicroRNAs can be classified as tumor suppressor or oncogenic. Loss of tumor suppressor and overexpression of oncogenic microRNAs are important events in regulating EMT in cancer. miRs-143 and -200 are frequently downregulated in several cancers. A possible mechanism for miR-143 downregulation includes the activation of KRAS and its downstream target Ras Responsive Element Binding Protein 1 (RREB1) [15]. Has-miR-200-3p is one of the fundamental EMT regulators and is known to target mesenchymal transcription factor ZEB1 [16].
2. ERK5 in Regulating the Metastatic Cascade and Therapeutic Interventions
The role of ERK5 in regulating the EMT axis is only recently being explored. Increasing evidence in the last decade suggests that ERK5 expression and activation have an essential role in regulating tumor characteristics that promote EMT and metastases, including morphological shift, degradation of the extracellular matrix, stemness and anchorage independent growth, angiogenesis, and immune evasion. The mechanisms that are responsible for the activation of ERK5 and subsequent EMT are outlined in Figure 2. All these events increase cell migration, invasion, and metastasis.
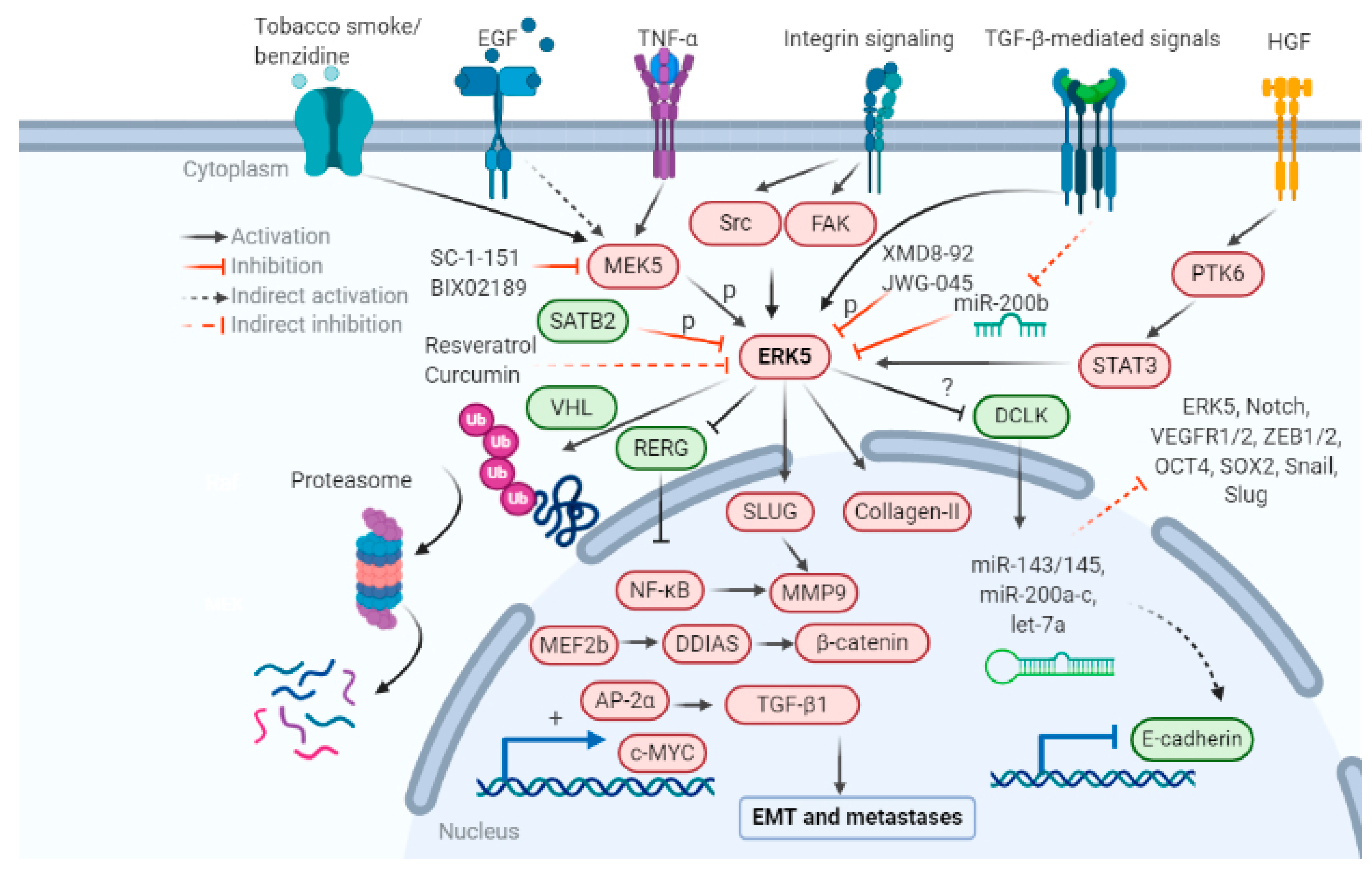
The role of the newest MAPK member ERK5 in various stages of the metastatic cascade are listed below: initiating with EMT, immune escape, increased production of collagen degrading enzymes, MMPs, collagenases, form and structure of the ECM, angiogenesis, survival in blood circulation, and secondary tumor growth at metastatic sites. Pharmacological inhibitors of the MEK5-ERK5 pathway SC-1-181, SC-1-151, XMD8-92, JWG-045, BIX02189, microRNAs -143, -200, and natural products resveratrol, curcumin, and vitamin D are a few candidates which can be used to target ERK5-mediated EMT in cancer research. Pharmacological interventions known to inhibit or reverse EMT and metastases are summarized in Table 2.
Table 2. Pharmacological interventions for targeting the ERK5 pathway to inhibit or reverse EMT.
| Pharmacological Intervention | Cancer | Target | Effect on EMT/MET |
|---|---|---|---|
| SC-1-151 | Triple-negative and tamoxifen-resistant breast cancer | MEK1/2 and MEK5 | EMT reversal |
| JWG-045 | Leukemia | ERK5 | Inhibition of EMT |
| XMD8-92 | Colon cancer, pancreatic cancer, breast cancer, leukemia, and lung cancer | ERK5 and BRD4 | Inhibition of EMT, stemness, and metastases |
| BIX02189 | Breast cancer, leukemia | MEK5 | Inhibition of TGFβ induced EMT |
| Resveratrol | Bladder cancer | STAT3/Twist-1 | Reversal of smoke induced EMT |
| Vitamin D | Acute myeloid leukemia | M-CSFR | Macrophage differentiation |
| Curcumin | Lung cancer, bladder cancer | ERK5/AP-1, ERK1/2, c-Fos, c-Jun, p-38 | Inhibition of smoke induced EMT |
| miR-143 | AML, Cholangiocarcinoma, and osteosarcoma | ERK5 | Inhibition of differentiation and apoptosis of AML cells; targets migration and invasion in osteosarcoma |
| miR-200 | Glioblastoma | ERK5, ZEB1 | EMT reversal |
In summary, SC-1-151, a dual MEK1/2 and MEK5 inhibitor, was found to reverse the mesenchymal phenotype, decrease mesenchymal markers, and increase epithelial marker E-cadherin in triple-negative and tamoxifen-resistant breast cancer [36]. XMD8-92, an ERK5 inhibitor, decreased stem-cell properties in colon cancer, inhibited EMT in PDAC, and attenuated inflammasome formation in malignant mesothelioma via downregulation of oncogenes c-MYC, KRAS, and VEGFR1/2, stemness regulators NOTCH1, OCT4, SOX2, and NANOG, EMT transcription factors ZEB1/2, SNAIL, and SLUG, and upregulation of tumor suppressor miRNAs -let-7a, -143/145, and -200 [20,84]. BIX02189, a MEK5 inhibitor, inhibited TGF-β induced EMT in breast cancer. Several natural products have been identified to attenuate EMT induced by benzidine. Resveratrol decreased smoke induced EMT in bladder cancer via STAT3/Twist1 inhibition [96]. Curcumin inhibited smoke-induced EMT as shown by an increase in the epithelial marker E-cadherin and zona occludens-1 (ZO-1) and a decrease in mesenchymal markers vimentin and N-cadherin in bladder cancer [97,98,99]. Vitamin D promoted the macrophagic differentiation of monocytes in acute myeloid leukemia (AML) via ERK5-mediated CSFR upregulation, thus promoting cancer cell phagocytosis. microRNA-143 or -200 restoration reversed EMT in osteosarcoma or glioblastoma, respectively, via ERK5 downregulation. These studies have demonstrated the relevance of targeting ERK5 to prevent or reverse EMT and metastases, thus validating ERK5 as an important therapeutic target in cancer.
Some of the ERK5 inhibitors were recently found to promote the nuclear translocation of ERK5 [100]. Hence, there is still a need for the discovery of effective ERK5 inhibitors/degraders. While these studies have been extremely helpful in identifying the less well-understood effects of ERK5 signaling via several mechanisms, there are several avenues that deserve more research in EMT and drug development; these include the cytosolic versus nuclear functions of ERK5 with respect to EMT, the role of total ERK5 versus phosphorylated ERK5, and its crosstalk with the immune system. The studies reviewed here show that ERK5 is a promising target in regulating EMT and metastases, and there is an urgent need to develop clinical drug candidates for targeting the ERK5 pathway.
This entry is adapted from the peer-reviewed paper 10.3390/biom11020183