Our understanding on the immunological roles of pathogen recognition in innate immunity has vastly increased over the past 20 years. Nucleotide-binding oligomerization domain (NOD)-like receptors (NLR) are cytosolic pattern recognition receptors (PRR) that are responsible for sensing microbial motifs and endogenous damage signals in mammalian cytosol for immune surveillance and host defense.
- allergy
- inflammasome
- molecular mechanism
- NOD-like receptors
- NLR
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Allergy is a chronic inflammatory condition triggered by allergens. Atopic dermatitis (AD) clinically characterized by pruritus, lichenification, reddening and dryness of the skin, and allergic asthma, featuring the presence of coughing, wheezing, tightness in the chest and shortness of breath, are two common allergic diseases found in children. The skin barrier and epithelial barrier of the lung not only provide a site of the entry for allergens, but also a route for pathogen invasion. Microbial and viral infections are common contributors to the symptoms and exacerbation in AD and allergic asthma. Gene–gene interaction, immune dysregulation as well as environmental factors are the keys in the pathogenesis of these two allergic diseases.
Innate immunity is the front line of defense against pathogens and dangers that is conserved in animals and plants. To initiate a rapid host defense and response against microbial infections, cellular damage and stress, intracellular and extracellular pattern recognition receptors (PRRs) will first recognize microbial signatures including Pathogen-Associated Molecular Patterns (PAMP), endogenous damage molecules Damage-Associated Molecular Patterns (DAMP), as well as stress signals. Unlike the adaptive immunity, this rapid recognition of pathogens and dangers by the germline-encoded PRRs does not require any priming or memory against pathogens that the cells encountered previously, but the highly conserved microbial patterns are vital for the survival of the microorganisms.
In response to the stimuli, the activated cytosolic PRRs will trigger protease caspase-1 to initiate the maturation and the subsequent release of inflammatory cytokines interleukin-1 (IL-1) and IL-18 or the induction of pyroptosis, which is a caspase 1-dependent programmed cell death that involves a rapid plasma-membrane rupture and pro-inflammatory content release of the microbial-infected or damaged cells [1]. These downstream inflammatory events ultimately facilitate the elimination of the invading pathogens; hence, they resolve infection and protect the host from disease development [2].
2. NLR Family
Major PRR families identified in mammals include Toll-like receptors (TLRs), nucleotide-binding leucine-rich repeat-containing proteins/nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid inducible gene I (RIG-I)-like receptors (RLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs), C-type lectin receptors (CLRs), pyrin and intracellular DNA sensors. Unlike the well-known PPR family TLRs and CLRs, which are transmembrane receptors that sense extracellular and endosomal microbes, NLRs belong to another large PPR family that is present in the cytosols and is responsible for non-specific recognition of intracellular microbial products, including peptidoglycan, which is a molecular complex present in all bacteria cell walls [3]. Moreover, NLR also serves as the backup host defense system against invaded pathogens and in tissues where other PRRs are compartmentalized and acts jointly with other PRRs, especially TLR, to generate a substantial innate immune response [4].
NLR is a protein structurally composed of three domains, namely (1) the microbial pattern recognition domain with leucine-rich repeats (LRR) located at the C terminus; (2) the nucleotide-binding NACHT-(NAIP (neural apoptosis inhibitory protein), CIITA (MHC Class II transactivator), HET-E (vegetative incompatibility protein from Podospora anserina) and TP-1 (telomerase-associated protein 1)) domain in the center that is essential for self-oligomerization, and (3) the terminal effector domain at the N terminus that drives downstream inflammatory caspase and NF-κB activation (Figure 1) [3]. There are so far around 22 NLR proteins identified, each carrying a distinct domain organization. Effector domains at the N terminus, which are crucial for signal transduction, help further divide these NLRs into five subfamilies, namely NLRA, NLRC, NLRC, NLRP and NLRX. Typical NLRs in the NLRC subfamily, which is characterized by the caspase activation and recruitment domain (CARD), include the nucleotide-binding oligomerization domain-containing protein 1 (NOD1) and NOD2; while the NLRP subfamily, which is characterized by the pyrin domain (PYD), includes NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) (Figure 1) [3]. NLRC and NLRP are the two major subfamilies that are found to be relevant to allergic diseases (Table 1).
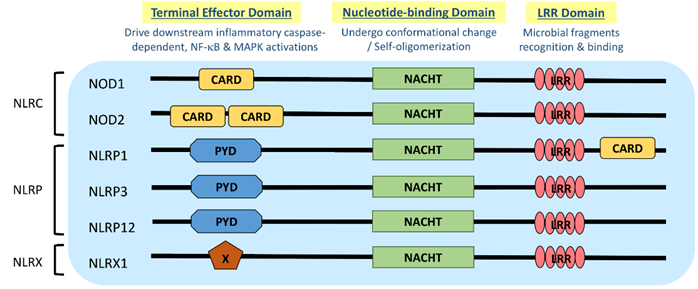
Figure 1. Domain architecture of NOD-like receptor (NLR): NOD1, NOD2, NLRP1, NLRP3, NLRP12 and NLRX1. NLR genes encode the intracellular multidomain proteins NLR, which are composed of a leucine-rich repeats (LRR) domain in the C terminal, the intermediate nucleotide-binding NACHT domain and a terminal effector domain in the N terminal. The variables caspase activation and recruitment domain (CARD), pyrin domain (PYD) and undefined domain (X) in the terminal effector domain further divide NLR into subfamilies, NLRC, NLRP and NLRX, respectively. Gene polymorphisms and gain-of-function mutations identified on these NLR genes in atopic dermatitis (AD) and allergic asthmatic patients may contribute to the aberrant expressions of NLR for microbial surveillance, asthma protection and exacerbation.
Table 1. Functional roles of NLRs in atopic dermatitis and allergic asthma.
|
Subfamily |
N-Terminal Domain |
NLR |
Functions Associated with AD |
Functions Associated with Allergic Asthma |
||
|
Rodents |
Human |
Rodents |
Human |
|||
|
NLRC |
CARD (Caspase recruitment domain) |
NOD1 (or CARD4) |
Detect γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP) from Gram-negative bacterial peptidoglycans [5]; |
Detect iE-DAP from Gram-negative bacterial peptidoglycans [5]; |
Expressed in mouse airway epithelial cells and some alveolar type II cells [8]; |
Expressed in human BEAS-2B cell and eosinophils [8,11], but not in basophils or KU812 cells [12]; |
|
|
|
NOD2 (or CARD15) |
Recognize MDP from nearly all bacteria and intracellular LPS [15]; |
Recognize muramyl dipeptide (MDP) from nearly all bacteria and intracellular LPS [15,18]; Synergize with TLR signaling pathway to initiate pro-inflammatory cytokines production [7] |
Recognize viral ssRNA genome of RSV, influenza virus and parainfluenza virus, and activate IRF3 and trigger IFN-γ production [21]; |
Expressed in human BEAS-2B cell, primary bronchial epithelial cells, eosinophils, basophils and KU812 cells [11,12,23]; |
|
NLRP |
PYD (Pyrin domain) |
NLRP1 (or NALP1) |
Nlrp1 is not expressed in keratinocytes * [25]; |
Detect MDP derived from bacterial peptidoglycan * [29]; |
|
May regulate asthma development and exacerbation [30] |
|
|
|
NLRP3 (or NALP3) |
Sense the cellular stress induced by a wide variety of stimuli, including extracellular ATP, pore-forming toxins, uric acid crystals, bacterial RNA, whole bacteria, synthetic purine-like compounds, viral DNA, peptidoglycans, UV radiation and reactive oxygen species; and hence trigger full activation of inflammasome [3,31–33]; |
Sense the cellular stress induced by a wide variety of stimuli, including extracellular ATP, pore-forming toxins, uric acid crystals, bacterial RNA, whole bacteria, synthetic purine-like compounds, viral DNA, peptidoglycans, UV radiation and reactive oxygen species; and hence trigger full activation of inflammasome [3,31]; |
NLRP3 are not involved in eosinophils regulation in allergic airway inflammation [44]; |
Not expressed in eosinophils [11]; |
|
|
|
NLRP12 |
Facilitate migration of BMDC from skin to draining lymph nodes [1]; |
|
Not involved in Th2-driven airway inflammation * [51] |
|
|
NLRX |
Unrelated domain |
NLRX1 (NLR family member X1) |
Regulate mitochondrial anti-viral response * [52]; |
|
Produce reactive oxidative species to enhance NF-κB and JNK pathways * [52]; |
Exposure to H. pylori can drive asthma protection [55] |
* Other potential functions related to AD/allergic asthma which are not discussed in the current review.
3. NLR and Atopy
Gene polymorphisms and haplotype combinations of NLR genes identified in atopic individuals revealed that NLR could be vital determinants of atopy susceptibility. NOD1 is located on the chromosome region 7q14-p15, which is a region believed to be atopy susceptible [6]. At least three of the polymorphisms located on the NOD and LLR domains of NOD1 gene are positively associated with the elevated total serum IgE level, while a haplotype on the NOD1 gene exerts a significant protective effect against IgE elevation in a German cohort [56]. Insertion polymorphism of the NOD2 gene may enhance the severity of atopic status, as it causes a 50% increase in risk of developing atopy and an elevated level in serum IgE in atopic children [57]. A Swedish cohort also revealed a significant association between the NLRP3 variant and total IgE antibody level exclusively in males [58]. The frameshift mutation may lead to truncation of the ligand-binding region LLR domain which then impairs the downstream NF-κB signaling upon bacterial lipopolysaccharide (LPS) stimulation [59]. Moreover, short isoform of NOD2/CARD15 was found to be an endogenous inhibitor of NOD2-induced NF-κB activation [60]. Impaired function of the innate immunity due to the CARD15 polymorphisms to recognize microbial exposure may give rise to Th2-dominant allergy [57]. The hygiene hypothesis suggested that early exposure to microbes can protect an individual from atopic disease by facilitating the development of the regulatory T cell (Treg) reservoir. Lack of the ability to recognize bacterial challenge may lead to the development of allergic diseases. It is also hypothesized that the NOD2 polymorphism affects the apoptosis of inflammatory cells, leading to a persistent and excessive inflammatory response as observed in atopic diseases, yet this hypothesis is yet to be proven [57].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22041507