Growing evidence suggests that the immune component of the tumor microenvironment (TME) in high-grade serous ovarian cancer (HGSOC) may play a significant role in the progression of the disease. The poor prognosis of HGSOC necessitates development of novel therapeutic strategies to improve patient outcomes. The type 2 diabetes medication, metformin, has been associated with significant improvement to overall survival in a number of retrospective clinical analyses. Recent data summarized here suggest that metformin may provide such a benefit through modulating the immune TME of HGSOC.
- ovarian cancer
- metformin
- omentum
- tumor microenvironment
1. Introduction
Metformin, a biguanide antidiabetic medication, has gained overwhelming attention in the treatment of inflammatory diseases, as well as a number of cancers. Early retrospective epidemiological case-control studies have indicated a possible association for metformin to enhance progression-free survival in patients with diabetes when compared to controls, which include patients with diabetes receiving other medications, as well as patients without diabetes [1][2]. Consistently, a subsequent meta-analysis of numerous retrospective studies showed a nearly twofold decrease in the risk of mortality across seven cohorts (cumulative odds ratio of 0.55), as well as a small but significant decrease in the incidence of ovarian cancer in several others [3].
In ovarian cancer, in vivo xenograft studies have shown that clinically relevant doses of metformin given in a preventative regimen, including pretreatment prior to and maintenance during engraftment, have decreased the size of the primary tumor and inhibited the number of metastatic implants [4], suggesting that the physiological effects of the drug may involve its activity in the TME. In line with these findings, recent studies have demonstrated that the drug could also specifically target mesothelial cells in a 3D organotypic model of invasion of the omentum, which was consistent with the decreased adhesion of HGSOC tumor cells to the omentum in explants removed from patients on metformin when compared to matched controls [5]. Moreover, in co-culture systems, metformin was observed to also inhibit chemoresistance by inhibiting NF-κB-dependent IL-6 secretion from fibroblasts[6], as well as adipocyte-induced tumor cell proliferation and migration [7].
2. Potential for Immunoregulation by Metformin in the Ovarian Tumor Microenvironment
2.1. Metformin and T Cells—An Overview
Evidence supports the claim that CD8+ TIL function is of great importance in the prognosis and the survival rate in patients with HGSOC. To date, the ability to pharmacologically modulate cytotoxic T cell behavior in the metastatic TME of HGSOC has not yet been evaluated using preclinical models. However, several studies demonstrate the potential impact of metformin on maintaining CD8+ TIL function and possibly enriching a high CD8+/CD4+ ratio to promote an immunoreactive environment in other cancers. In a study by Eikawa et al. [8], metformin demonstrated inhibition of the growth of solid tumors in vivo in leukemia, melanoma, non-small-cell lung carcinoma, breast cancer, renal cancer, and intestinal cancer due to its ability to halt the exhaustion and apoptosis of CD8+ TILs in the TME. These findings were exemplified through the increase of three cytokines that the PD1−/Tim3+ phenotypic CD8+ TILs produce, consisting of interleukin-2 (IL-2), tumor necrosis factor alpha (TNFα), and interferon gamma (IFNγ). They also illustrated how metformin induced the differentiation from central memory T cells (Tmem) to effector memory T cells, demonstrating a stronger memory immune response to recurring tumor development. The authors attributed these findings in part to 5′-AMP-activated protein kinase (AMPK)-dependent inhibition of the mammalian target of rapamycin (mTOR) [8]. More recently, Kunisada et al. observed that metformin inhibited Treg induction and activity at the site of tumors in fibrosarcoma, leukemia, and melanoma in vitro by downregulating Foxp3, a transcription factor that promotes Treg phenotypes, thereby hindering the TGF-β-dependent differentiation of naïve CD4+/CD25− T cells into Tregs [9]. Subsequent studies evaluated metformin’s effect on the ratio between tumor-infiltrating Tregs and tumor-infiltrating CD8+ T cells. When treated with metformin, an increase in CD8+ TILs was observed, suggesting that metformin promoted either increased recruitment or differentiation toward an antitumor T cell phenotype. Moreover, in vivo findings showed that metformin effectively delayed tumor growth by causing a shift from oxidative phosphorylation (OXPHOS) to glycolysis, which then decreased the Treg expression of interleukin-10 (IL-10) and cytotoxic T lymphocyte antigen-4 (CTLA-4) [9]. Intriguingly, the ability of metformin to alter CD4+ T cell phenotypes in vivo was associated with its activation of mTOR, as the mTOR inhibitor rapamycin ablated the ability of metformin to suppress Treg abundance. This mechanism is in contrast with the observed prevention of CD8+ TIL exhaustion resulting from the inhibition of mTOR by AMPK in the previous study by Eikawa et al. While convincing, it is contradictory to its canonical negative regulation of mTOR that was observed in most reports across disease states (reviewed in [10]). Taken together, these data suggest that metformin may have unique effects on specific subtypes of T cells, possibly due to their distinctive metabolic features [11], as well as their markedly different proteomic responses to external stimuli [12]. However, both indicate that metformin may favor an immunoreactive distribution of T cell phenotypes. The potential regulation of T cell differentiation and the subsequent CTC/Treg ratio by metformin is depicted in Figure 1.
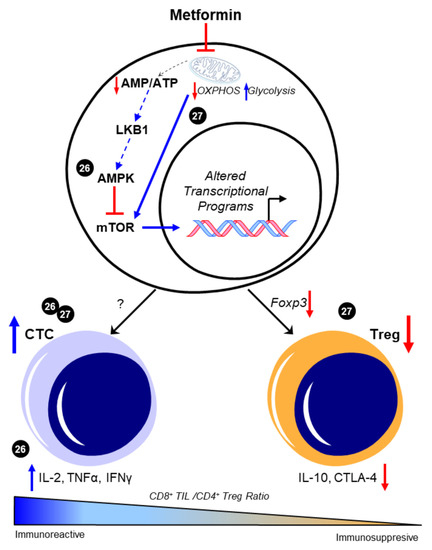
Figure 1. Metformin enhances CD8+/CD4+ ratio to favor an immunoreactive microenvironment. The canonical pathway of metformin intracellular activity that results in 5′-AMP-activated protein kinase (AMPK)-dependent inhibition of the mammalian target of rapamycin (mTOR) is shown. Presumably, this enhancement in AMPK and the consequent mTOR inhibition is attributed to the metabolic effects of metformin in promoting glycolysis through inhibited mitochondrial respiration; however, this was not experimentally shown in these studies and is represented by dotted lines. The ability of metformin to increase the CD8+/CD4+ ratio [8][9] was associated with increases in functional CD8+ tumor-infiltrating lymphocytes (TILs) and concomitant increases in interleukin 2 (IL-2), tumor necrosis factor alpha (TNFα), and interferon gamma (IFNγ) [8]. Decreases in Foxp3-driven CD4+ T cell phenotypes and the subsequent reduction in interleukin 10 (IL-10) and cytotoxic T lymphocyte antigen-4 (CTLA-4) expression were also separately observed [9]. Cited experiments are denoted by their respective reference numbers [8][9]. CTC: cytotoxic CD8+ TILs, OXPHOS: oxidative phosphorylation, Treg: regulatory T cells. Blue arrows indicate upregulation/activation, red arrows indicate downregulation/inactivation and red T symbols indicate direct inhibition. Solid lines represent data presented in the cited manuscript; dotted lines indicate informed interactions from well-established data.
2.2. Metformin and T Cells—Metabolic Targets and T Cell Phenotypes
The potential influence of AMPK-dependent metabolic reprogramming on T cell phenotypes has recently been reviewed [13], and as it is a direct substrate for LKB1 [14], may provide further insight into possible mechanisms by which metformin may regulate T cell differentiation and function. The exact relationship between metformin and LKB1, AMPK, and mTOR in T cell differentiation and function is unclear. Notably, metformin has not been shown to directly interact with either LKB1 or AMPK and may indeed have multiple intracellular targets in addition to its putative binding to mitochondrial complex I [15][16][17]. This may be especially relevant in its potential to directly bind and inhibit HMGB1 [16], a cytokine that is observed to regulate T cell activity and have a multitude of effects on other immune cells (reviewed in [18]). How the inhibition of HMGB1 by metformin could impact T cell function in the complex environment of the TME has yet to be evaluated. There is also some contention as to whether the doses exceeding the micromolar concentrations used in in vitro studies, including some of the studies discussed in this review, are relevant to the exposure of patients on the drug at doses indicated for its use in managing diabetes, which ranges from 500–2000 mg per day. However, it is suggested that treatment regimens often used in vivo are more closely representative of the concentration of the drug that would accumulate in target tissues, and which activate AMPK [19]. The possibility that metformin is acting independently of LKB1/AMPK in T cells in vivo may explain some of these discrepancies regarding the drug’s observed physiological activity and what has been demonstrated using genetic mouse models directly targeting the LKB1/AMPK axis. In addition to its possible effects on multiple cell types within the TME, a further complication arises in the systemic effects of metformin that may impact T cell function, namely, its antihyperglycemic effects through enhanced insulin sensitivity, which could impact glucose-dependent T cell differentiation and activity [20][21]. Thus, a more comprehensive understanding of the exact molecular mechanisms by which metformin regulates T cell development and functions in vivo, or in physiologically relevant in vitro models of the TME, are specifically required in order to properly assess the direct effects of the drug on T cell activity in the TME.
2.3. Metformin and Myeloid-Derived Suppressor Cells (MDSCs)—An Overview
The use of compound C, an AMPK inhibitor, and CoCl2, an HIF1α activator, warrants caution with the interpretation of the results mentioned above due to the non-specificity of both drugs in their intracellular activity (e.g., [22][23]). However, these findings are consistent with the reported inhibition in MDSC function by AMPK activation and the proposed involvement of AMPK in the metabolic regulation of MDSC activity [24][25], as well as the putative transcriptional regulation of CD39 and CD73 by HIF1α [26][27]. Further testing by Li et al. showed that metformin treatment increased the production of granzyme B, perforin, and IFNγ by CD8+ T cells in vitro and in vivo, which was associated with decreased activity of immunosuppressive MDSCs resulting from the downregulation of CD39 and CD73 [28], as illustrated in Figure 2.
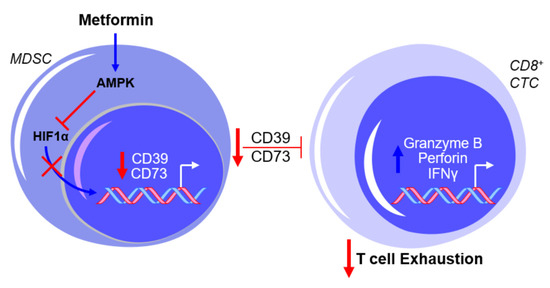
Figure 2. Metformin prevents myeloid-derived suppressor cell (MDSC)-mediated T cell exhaustion by downregulating CD39 and CD73 expression. The exposure of MDSCs to metformin activates AMPK and inhibits hypoxia-inducible factor 1-alpha (HIF1α) expression, thereby inhibiting the HIF1α-mediated transcriptional upregulation of CD39 and CD73 and subsequently suppressing cytotoxic CD8+ T lymphocytes (CTCs). This results in a net effect of reduced T cell exhaustion. Blue arrows indicate upregulation/activation, red arrows indicate downregulation/inactivation and red T symbols indicate inhibition.
2.4. Metformin and MDSCs—Clinical Implications and Limitations
To complement their preclinical findings, the level of expression of CD39 and CD73 in MDSCs, as well as CD8+ T cell function, was measured in patients with type 2 diabetes mellitus (T2DM) on metformin. Consistent with their in vitro and in vivo data, metformin treatment significantly decreased CD39 and CD73 expression in MDSCs but increased CD8+ T cell function, as measured using granzyme B production. Intriguingly, these effects were observed in patients with diabetes before and after metformin treatment using a pairwise comparison (e.g., comparing the expression levels within patients following treatment) [28]. Presumably, these measurements were taken at the beginning and end of the 2-year prospective study; however, the exact duration and dosage of treatment is not specified. Nonetheless, the concept that 2 years (or less) of metformin treatment could have a profound impact on MDSC behavior in the TME is a strong indication that the drug may be useful in intervention treatment regimens, especially in patients that exhibit an immunosuppressed TME, or potentially as a chemopreventive agent in patients who are identified as being at high risk for developing an aggressive disease.
Taken together, this study by Li et al. supports a possible mechanism for metformin that prevents T cell exhaustion through inhibition by other immune cells, such as MDSCs, to maintain an immunoreactive TME. While promising, the in vitro data reached significance only at supraphysiologic levels of metformin at 2mM, which is markedly higher than the micromolar range (<100 µM) experimental equivalent to the clinically used doses of the drug [19]. It is unclear whether higher doses of the drug were necessary due, at least in part, to the high levels of glucose in the media during the metformin exposure, as its biological activity in vitro (e.g., AMPK activation) has been demonstrated to be significantly restricted by the elevated glucose observed in most cell culture media [29]. The ex vivo analysis of T cells from mice exposed to the drug also markedly exceeded clinically relevant concentrations at 10 mM; however, these data were strongly supported by the clinical findings. Further investigation on metformin’s modulation of the regulation of the immune milieu in the TME by MDSCs continues to be essential to determine whether metformin may be utilized as a therapeutic strategy to prevent immune evasion and the subsequent progression of HGSOC.
2.5. Metformin and Neutrophils—Neutrophil:Lymphocyte Ratio (NLR)
There is no clear indication that metformin may impact the NLR in patients with HGSOC. However, data in other inflammatory disease states suggest that the drug could potentially reduce NLR in several contexts. When looking at metformin in diabetes, a disease that has been observed to have an elevated NLR [30], it was observed that T2DM patients taking metformin had significantly decreased mean NLRs when compared to patients being treated with a sulfonylurea [31]. Patients with polycystic ovarian syndrome (PCOS), a disease associated with chronic inflammatory states and that has been associated with up to a threefold increased risk of developing OvCa [32][33], also often present with an elevated leukocyte count, which is largely due to the increased levels of circulating neutrophils [34][35]. When Ibanez and colleagues used metformin as a treatment compared to a placebo, metformin lessened the inflammatory response by significantly reducing the neutrophil count within 3 months of treatment [35]. Although these data were obtained from patients with hyperinsulinemic hyperandrogenism, a hallmark of PCOS in which patients display especially high neutrophil and leukocyte counts [35], it does support the possibility that metformin could prevent the increased neutrophil levels that are associated with aggressive HGSOC [36].
2.6. Metformin and Neutrophils—Neutrophil Extracellular Trap (NET)
Excess neutrophil count could predictably lead to increased formation of neutrophil extracellular traps (NETs), which are the secretion of a network of fibers including chromatin and proteins that generally have microbicidal activity (reviewed in [37]). NET formation may also result in a specific form of cell death referred to as NETosis, in which destabilization of neutrophil membranes causes the release of a dense network of NETs that may induce persistent inflammation associated with several autoimmune diseases [37]. In a recent report by Honami Naora’s group [38] it was suggested that the increase in neutrophils migrating into the omentum facilitated the implantation of OvCa at this metastatic site. This resulted in detrimental effects of OvCa-induced inflammatory signaling stimulating NET formation and subsequent NETosis. Notably, it was found that OvCa cells attached to NETs to metastasize into the omentum. If NET extrusion was inhibited by genetic knockdown of a necessary enzyme for their formation (Peptidylarginine Deiminase 4 (PAD4)), omental metastasis was essentially ablated. There was no apparent effect of inhibited NET formation on development of the primary ovarian tumor, suggesting that NETs promote early dissemination and implantation of OvCa cells onto the omentum [38]. This study exemplifies the effectiveness of neutrophil depletion to prevent metastasis in early-stage OvCa, and strengthens the argument that the use of an agent, such as metformin, in a chemoprevention strategy in patients at high risk could potentially prevent early metastasis and improve patient outcomes. To this effect, studies by Menegazzo et al. identified the beneficial impact of metformin on NETs and NETosis [39]. In a randomized controlled trial, metformin was shown to significantly decrease concentrations of NET biomarkers, such as dsDNA, histones, neutrophil elastase, and proteinase-3.
While there is no direct evidence in HGSOC, these studies suggest that metformin’s anti-inflammatory effects may include a reduced neutrophil count, a subsequent decreased NLR, and the inhibition of NETosis (summarized in Figure 3), all of which have been identified as independent factors that are associated with decreased progression-free survival in OvCa. These data warrant further investigation of metformin and its regulation of NLR and NETosis in OvCa. By expanding in vivo studies to assess circulating neutrophils and leukocytes and fully characterizing the distribution of various immune cells within the primary tumor and secondary sites, as well as determining the impact of metformin on neutrophil functions (i.e., NETosis) in the omental TME, it will be possible to evaluate the effects of metformin on preventing a deleteriously pro-inflammatory microenvironment that promotes the metastasis of HGSOC.
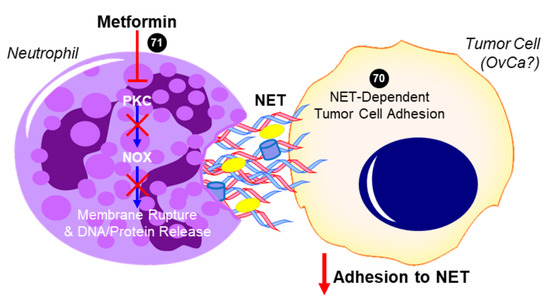
Figure 3. Proposed role of metformin in inhibiting neutrophil extracellular trap (NET)-induced tumor cell metastasis. In addition to decreasing neutrophil recruitment (not shown), metformin has been shown to inhibit protein kinase C beta II (PKCβII) translocation to the plasma membrane and prevent the downstream activation of NADPH oxidase (NOX) that would otherwise result in the expulsion of neutrophil extracellular trap (NET) components into the extracellular space [39]. Tumor cells have been shown to adhere to NETs, which contain DNA, histones, neutrophil elastase, and other proteins. In the case of ovarian cancer (OvCa), NET formation was especially critical to the ability of tumor cells to colonize the omentum in vivo, while having no effect on the primary tumor or other intra-abdominal sites [38]. The ability for metformin to inhibit this NET-dependent adhesion and colonization of the omentum has not yet been investigated but is a potential mechanism for its antitumor activity. Cited experiments are denoted by their respective reference numbers [38][39]. Blue arrows indicate activation, red arrows indicate inhibition, red T symbols indicate direct inactivation, and red X symbols indicate inhibition of this pathway by metformin.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22020867
References
- Romero, I.L.; McCormick, A.; McEwen, K.A.; Park, S.; Karrison, T.; Yamada, S.D.; Pannain, S.; Lengyel, E. Relationship of Type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet. Gynecol. 2012, 119, 61–67.
- Kumar, S.; Meuter, A.; Thapa, P.; Langstraat, C.; Giri, S.; Chien, J.; Rattan, R.; Cliby, W.A.; Shridhar, V. Metformin intake is associated with better survival in ovarian cancer: A case-control study. Cancer 2013, 119, 555–562.
- Shi, J.; Liu, B.; Wang, H.; Zhang, T.; Yang, L. Association of metformin use with ovarian cancer incidence and prognosis: A systematic review and meta-analysis. Int. J. Gynecol. Cancer 2019, 29, 140–146.
- Lengyel, E.; Litchfield, L.M.; Mitra, A.K.; Nieman, K.M.; Mukherjee, A.; Zhang, Y.; Johnson, A.; Bradaric, M.J.; Lee, W.; Romero, I.L. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am. J. Obstet. Gynecol. 2015, 212, 479.e1–479.e10.
- Hart, P.C.; Kenny, H.A.; Grassl, N.; Watters, K.M.; Litchfield, L.M.; Coscia, F.; Blaženović, I.; Ploetzky, L.; Fiehn, O.; Mann, M.; et al. Mesothelial Cell HIF1α expression is metabolically downregulated by metformin to prevent oncogenic tumor-stromal crosstalk. Cell Rep. 2019, 29, 4086–4098.
- Tebbe, C.; Chhina, J.; Dar, S.A.; Sarigiannis, K.; Giri, S.; Munkarah, A.R.; Rattan, R. Metformin limits the adipocyte tumor-promoting effect on ovarian cancer. Oncotarget 2014, 5, 4746–4764.
- Xu, S.; Yang, Z.-Y.; Jin, P.; Yang, X.; Li, X.; Wei, X.; Wang, Y.; Long, S.; Zhang, T.; Chen, G.; et al. Metformin suppresses tumor progression by inactivating stromal fibroblasts in ovarian cancer. Mol. Cancer Ther. 2018, 17, 1291–1302.
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814.
- Kunisada, Y.; Eikawa, S.; Tomonobu, N.; Domae, S.; Uehara, T.; Hori, S.; Furusawa, Y.; Hase, K.; Sasaki, A.; Udono, H. Attenuation of CD4 + CD25 + Regulatory T cells in the tumor microenvironment by metformin, a Type 2 diabetes drug. EBioMedicine 2017, 25, 154–164.
- Amin, S.; Lux, A.; O’Callaghan, F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019, 85, 37–46.
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.; Opipari, A.W., Jr.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115.
- Gerner, M.C.; Niederstaetter, L.; Ziegler, L.; Bileck, A.; Slany, A.; Janker, L.; Schmidt, R.L.J.; Gerner, C.; Del Favero, G.; Schmetterer, K.G. Proteome analysis reveals distinct mitochondrial functions linked to interferon response patterns in activated CD4+ and CD8+ T Cells. Front. Pharmacol. 2019, 10, 727.
- Ma, E.H.; Poffenberger, M.C.; Wong, A.H.; Jones, R.G. The role of AMPK in T cell metabolism and function. Curr. Opin. Immunol. 2017, 46, 45–52.
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; Depinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335.
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228.
- Horiuchi, T.; Sakata, N.; Narumi, Y.; Kimura, T.; Hayashi, T.; Nagano, K.; Liu, K.; Nishibori, M.; Tsukita, S.; Yamada, T.; et al. Metformin directly binds the alarmin HMGB1 and inhibits its proinflammatory activity. J. Biol. Chem. 2017, 292, 8436–8446.
- Kinsky, O.R.; Hargraves, T.L.; Anumol, T.; Jacobsen, N.E.; Dai, J.; Snyder, S.A.; Monks, T.J.; Lau, S.S. Metformin scavenges methylglyoxal to form a novel imidazolinone metabolite in humans. Chem. Res. Toxicol. 2016, 29, 227–234.
- Li, G.; Liang, X.; Lotze, M.T. HMGB1: The central cytokine for all lymphoid cells. Front. Immunol. 2013, 4, 68.
- Chandel, N.S.; Avizonis, D.; Reczek, C.R.; Weinberg, S.E.; Menz, S.; Neuhaus, R.; Christian, S.; Haegebarth, A.; Algire, C.; Pollak, M. Are metformin doses used in murine cancer models clinically relevant? Cell Metab. 2016, 23, 569–570.
- Palmer, C.S.; Ostrowski, M.; Balderson, B.; Christian, N.; Crowe, S.M. Glucose metabolism regulates T cell activation, differentiation, and functions. Front. Immunol. 2015, 6, 1.
- Tsai, S.; Clemente-Casares, X.; Zhou, A.C.; Lei, H.; Ahn, J.J.; Chan, Y.T.; Choi, O.; Luck, H.; Woo, M.; Dunn, S.E.; et al. Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab. 2018, 28, 922–934.
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2007, 4, 33–41.
- Li, Q.; Ke, Q.; Costa, M. Alterations of histone modifications by cobalt compounds. Carcinogenesis 2009, 30, 1243–1251.
- Trikha, P.; Plews, R.L.; Stiff, A.; Gautam, S.; Hsu, V.; Abood, D.; Wesolowski, R.; Landi, I.; Mo, X.; Phay, J.; et al. Targeting myeloid-derived suppressor cells using a novel adenosine monophosphate-activated protein kinase (AMPK) activator. OncoImmunology 2016, 5, e1214787.
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK activation inhibits the functions of myeloid-derived suppressor cells (MDSC): Impact on cancer and aging. J. Mol. Med. 2019, 97, 1049–1064.
- Sarkar, K.; Cai, Z.; Gupta, R.; Parajuli, N.; Fox-Talbot, K.; Darshan, M.S.; Gonzalez, F.J.; Semenza, G.L. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2012, 109, 10504–10509.
- Kling, L.; Benck, U.; Breedijk, A.; Leikeim, L.; Heitzmann, M.; Porubsky, S.; Krämer, B.K.; Yard, B.A.; Kälsch, A.-I. Changes in CD73, CD39 and CD26 expression on T-lymphocytes of ANCA-associated vasculitis patients suggest impairment in adenosine generation and turn-over. Sci. Rep. 2017, 7, 11683.
- Li, L.; Wang, L.; Li, J.; Fan, Z.; Yang, L.; Zhang, Z.; Zhang, C.; Yue, D.; Qin, G.; Zhang, T.; et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018, 78, 1779–1791.
- Litchfield, L.M.; Mukherjee, A.; Eckert, M.A.; Johnson, A.; Mills, K.A.; Pan, S.; Shridhar, V.; Lengyel, E.; Romero, I.L. Hyperglycemia-induced metabolic compensation inhibits metformin sensitivity in ovarian cancer. Oncotarget 2015, 6, 23548–23560.
- Duman, T.T.; Aktas, G.; Atak, B.M.; Kocak, M.Z.; Erkus, E.; Savli, H. Neutrophil to lymphocyte ratio as an indicative of diabetic control level in type 2 diabetes mellitus. Afr. Health Sci. 2019, 19, 1602–1606.
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ. Res. 2016, 119, 652–665.
- Schildkraut, J.M.; Schwingl, P.J.; Bastos, E.; Evanoff, A.; Hughes, C. Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet. Gynecol. 1996, 88, 554–559.
- Kelly, C.C.J.; Lyall, H.; Petrie, J.R.; Gould, G.W.; Connell, J.M.; Sattar, N. Low grade chronic inflammation in women with polycystic ovarian syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 2453–2455.
- Orio, F.; Palomba, S.; Cascella, T.; Di Biase, S.; Manguso, F.; Tauchmanovà, L.; Nardo, L.G.; Labella, D.; Savastano, S.; Russo, T.; et al. The increase of leukocytes as a new putative marker of low-grade chronic inflammation and early cardiovascular risk in polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 2–5.
- Ibáñez, L.; Jaramillo, A.M.; Ferrer, A.; de Zegher, F. High neutrophil count in girls and women with hyperinsulinaemic hyperandrogenism: Normalization with metformin and flutamide overcomes the aggravation by oral contraception. Hum. Reprod. 2005, 20, 2457–2462.
- Yin, X.; Wu, L.; Yang, H.; Yang, H. Prognostic significance of neutrophil–lymphocyte ratio (NLR) in patients with ovarian cancer. Medicine 2019, 98, e17475.
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147.
- Lee, W.; Ko, S.Y.; Mohamed, M.S.; Kenny, H.A.; Lengyel, E.; Naora, H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J. Exp. Med. 2019, 216, 176–194.
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.V.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601.