Immunometabolism is a relatively new field of research that aims at understanding interconnections between the immune system and cellular metabolism. This is now well-documented for innate immune cells of the myeloid lineage such as macrophages and myeloid dendritic cells (DCs) when they engage their differentiation or activation programs. Several studies have shown that stimulation of DCs or macrophages by the binding of pathogen-associated molecular patterns (PAMPs) to pattern recognition receptors (PRRs) leads to increased glycolytic activity and rewiring of central carbon metabolism. These metabolic modulations are essential to support and settle immunological functions by providing energy and immunoregulatory metabolites. As the understanding of molecular mechanisms progressed, significant differences between cell types and species have also been discovered. Pathways leading to the regulation of central carbon metabolism in macrophages and DCs by PRR signaling and consequences on cellular functions are reviewed here.
- innate immunity
- dendritic cells
- macrophages
- toll-like receptor (TLR) signaling
- Immunometabolism
- central carbon metabolism
- glycolysis
- TCA cycle
- oxidative phosphorylation
- lipid metabolism
1. Mechanisms Controlling Glycolytic Reprogramming in Myeloid Cells
The molecular mechanisms controlling glycolytic reprogramming upon TLR stimulation have been partially uncovered and differ according to cell types. In murine BMDCs stimulated through TLR4 by LPS, the early increase in glycolysis is controlled by activation of TBK1, IKKε and AKT kinases, favoring mitochondrial translocation of HK2[1] (Figure 1). Relocalization of HK2 to the mitochondria, occurring within the first hour of LPS stimulation, enhances HK activity, thus participating in the early glycolytic burst, fueling the TCA cycle and fatty acids (FA) synthesis. Glycogen metabolism supports early glycolytic reprogramming required for DC immune responses[2][3]. The modulation of pyruvate kinase M2 (PKM2) activity, another key enzyme of glycolysis, is required to induce inflammatory responses in macrophages. Deacetylation of PKM2 by class II histone deacetylases enhances its activity and thus promotes LPS-inducible interleukin (IL)-1β production in human and mouse macrophages[4]. Upon TLR4 stimulation, the expression of inducible nitric oxide synthase (iNOS) is also upregulated in mouse macrophages and DCs. This enzyme catalyzes the production of nitrogen oxide (NO) from L-arginine. NO, that diffuses in the microenvironment, then interferes with TCA cycle functioning and inhibits mitochondrial electron transport chain complexes, thus reducing O2 consumption and ATP production by OXPHOS[5]. The late increase in glycolytic metabolism observed in these cells may be a survival mechanism to maintain ATP production despite OXPHOS inhibition[6]. Importantly, human macrophages, which do not produce significant NO, present unaltered mitochondrial respiration upon LPS stimulation[7][8].
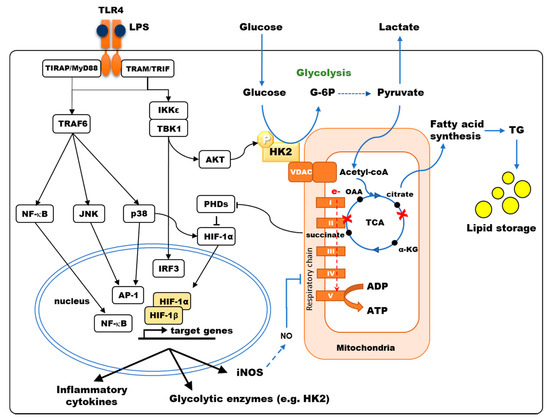
Figure 1. TLR4 signaling modulates the central carbon metabolism of macrophages and myeloid dendritic cells. TLR4 stimulation results in the activation of NF-κB, JNK, p38-MAPK, inducing the secretion of pro-inflammatory cytokines, and of TBK1/IKKε inducing IRF3-dependent type I interferon and AKT phosphorylation, favoring Hexokinase-2 (HK2) binding to VDAC. Hexokinase (HK) activity is the rate-limiting enzyme controlling glucose entry into glycolysis. Pyruvate is converted to acetyl-CoA into the mitochondria, fueling the TCA cycle. LPS stimulation of murine bone-marrow-derived DCs and macrophages results in a broken TCA cycle (red crosses), where succinate accumulates, inhibiting prolyl-hydroxylase domain enzymes (PHDs) thus favoring HIF-1α accumulation, and citrate is diverted from the TCA cycle to fuel fatty acid synthesis. HIF-1α induces the transcription of genes such as glycolytic enzymes, inducible nitric oxide synthase (iNOS) and pro-IL-1β. In human monocyte-derived DCs, p38-MAPK activation results in HIF-1α accumulation, enhancing the expression of metabolic enzymes such as HK2. Under aerobic conditions, electron (e-) transport through the respiratory chain (Complex I to V) generates ATP by oxidative phosphorylation (OXPHOS). In murine DCs, OXPHOS is inhibited by NO production upon LPS stimulation.
Hypoxia-inducible factor 1-alpha (HIF-1α) is a master transcriptional regulator of glycolytic enzymes, including HK and PKM2[9]. Under normoxia, HIF-1α degradation is induced by prolyl-hydroxylase domain enzymes (PHDs) that hydroxylate proline and asparagine residues. The interaction of the von Hippel–Lindau (VHL) factor with hydroxylated residues of HIF-1α recruits another component of an E3 ubiquitin–ligase that targets this factor to the proteasome for degradation[10]. PHD requires O2 and α-ketoglutarate (α-KG) to hydroxylate HIF-1α and generates CO2 and succinate as byproducts of this reaction. Hence, PHD activity is controlled by intracellular concentrations of O2 and α-KG as substrates but is also regulated by succinate, which is acting as a competitive inhibitor of these enzymes. Hypoxia inhibits PHDs, resulting in HIF-1α accumulation[11]. Nevertheless, in normoxic conditions, alternative mechanisms also result in HIF-1α accumulation upon LPS stimulation. PHD inhibition depends on reactive oxygen species (ROS) production and succinate accumulation, thus increasing HIF-1α stability [12][13]. In murine macrophages and DCs stimulated by LPS, the signaling of mammalian target of rapamycin complex 1 (mTORC1), whose activation depends on available nutrients including glucose, is sustained, and HIF-1α is upregulated, thus increasing glycolysis and triggering iNOS expression with consequences on OXPHOS as aforementioned[14][15][16]. Moon et al. show that Raptor/mTORC1 complex is involved in the regulation of HK1 expression and glycolysis that regulates NLRP3 inflammasome activation[15]. This glucose-sensitive signal transduction circuit coordinates DC metabolism and function to limit DC-stimulated T-cell responses[16]. In glucose-deprived cells, this mTORC1/HIF-1α/iNOS pathway is impaired, thus impacting both DC metabolism and immunological functions[17]. In these cells, NF-κB- and ERK-dependent transcriptional events, which are induced upon TLR engagement, are also required to trigger HIF-1α accumulation[18][19]. mTORC2 also enhances glycolytic metabolism by activating AKT and promoting MYC transcription activation[20]. In addition, PKM2 can associate and regulate HIF-1α activity with consequences on IL-1β induction in LPS-activated macrophages[21]. We have shown that quite differently, the increased expression of HIF-1α observed in human MoDCs stimulated by TLR4 depends on p38-MAPK activation[22]. HIF-1α then increases HK2 levels in both human MoDCs and mouse BMDCs, resulting in higher HK activity and glycolytic flux[22][1]. In human MoDCs, cytokine secretion triggered by TLR4 stimulation depends on this p38-MAPK/HIF-1α/HK2 pathway, while other pathways are controlling the induction of maturation markers such as MHC-class II, CD40, and CD86[22]. Although TLR1/2 or TLR2/6 stimulation also results in a glycolytic burst in human MoDCs, the molecular mechanisms involved are not dependent on p38-MAPK activation of HIF-1α[22]. Beyond its impact on glycolysis, this upregulation of HK2 has consequences on apoptosis and autophagy due to the nonenzymatic functions of this protein. Overexpression and mitochondrial association of HK2 confer protection to apoptotic or necrotic stimuli in different cell types by several mechanisms[23]. Moreover, in response to glucose starvation, HK2 binds and inhibits mTORC1, thus facilitating autophagy[23]. Although established in non-immune cells, this mechanism could contribute to protecting DCs or macrophages from cellular damage, providing energy by recycling intracellular components, but also contribute to internalized pathogen processing, antigen presentation and immune activation.
2. TCA Rewiring
Metabolic reprogramming by TLR stimulation is not limited to enhanced glycolysis but also results in reconfiguration of the TCA cycle, which contributes to macrophage and DC activation[24]. TLR4 activation results in the inhibition of pyruvate dehydrogenase (PDH) in murine macrophages, thus limiting the metabolization of pyruvate into acetyl-CoA, which is normally fueling the TCA cycle (Figure 1)[19]. This restriction of pyruvate entry into the TCA is a late event occurring after LPS + IFNɣ stimulation[25]. LPS stimulation of murine macrophages also leads to reduced expression of several enzymes of the TCA cycle, therefore modifying the balance of flux for different metabolites[19]. For example, decreased expression of iso-citrate dehydrogenase (IDH) results in citrate accumulation in M1 macrophages[17]. When macrophages are stimulated by LPS, IRG1 is upregulated, favoring the production of itaconate from citrate via aconitate. Itaconate, by reducing the production of IL-1β, IL-12, IL-6 and ROS triggered by LPS stimulation, exhibits anti-inflammatory effects[26][27]. It was recently shown that itaconate could induce electrophilic stress, promoting Nrf2-dependent transcription and altering cellular response to TLR via the inhibition of IκBζ/ATF3-mediated inflammation, reducing IL-6 and IL-12 secretion[28]. Membrane permeable derivatives of itaconate have revealed potent anti-inflammatory activities that could limit excessive innate immunity induced by human pathogenic viruses, including SARS-CoV-2[29]. Itaconate is also an inhibitor of succinate dehydrogenase (SDH; complex-II of the electron transport chain). SDH inhibition by itaconate blocks the TCA cycle resulting in succinate accumulation, thus linking citrate and succinate increase. As opposed to itaconate, succinate is acting as a pro-inflammatory signal in murine macrophages. When the electron transfer chain is inhibited downstream of complex-II, succinate oxidation provides electrons to complex I (reverse electron transport). This generates ROS leading to increased expression of pro-IL-1β and activation of the NLRP3 inflammasome for processing pro-IL-1β into IL-1β[30]. Consequently, the inhibition of complex I by metformin prevents the production of ROS and IL-1β secretion by LPS-stimulated cells[31]. Moreover, succinate accumulation inhibits prolyl-hydroxylases (PHDs), thus promoting HIF-1α accumulation and inducing the expression of glycolytic enzymes[9] and inflammatory cytokines such as IL-1β[10] without affecting TNFα secretion [10][30]. Thus, itaconate and succinate may have balanced effects on inflammation depending on the kinetics of production and the half-life of these metabolites.
Because of this broken TCA cycle, replenishment of intermediary metabolites is necessary. In particular, α-KG and succinate are generated from glutamine by glutamine dehydrogenase and the γ-aminobutyric acid (GABA) shunt. Succinate is also a secreted metabolite that is signaling in an autocrine and paracrine manner. Succinate is a chemotactic factor enhancing the activation of DCs stimulated with TLR ligands[32]. In addition, engagement of its membrane receptor SUCNR1 (also known as GPR91) triggers IL-1β production[33]. In pathological conditions, such as rheumatoid arthritis and obesity, cell signaling downstream of SUCNR1 was shown to promote inflammation in myeloid cells[33][34]. However, SUCNR1 can also favor the M2 polarization of macrophages, thus limiting inflammation in obesity [35].
3. Lipid Metabolism
Citrate efflux from the mitochondria is the main source of carbon for FA synthesis and generates inflammatory free radicals (Figure 2). In murine M1 macrophages, reduced IDH expression inhibits the TCA cycle by preventing citrate degradation. As a result, citrate efflux to the cytosol by the specific transporter SLC25A1 is increased[17]. LPS stimulation in macrophages and BMDCs has been shown to induce the expression of this transporter, further increasing citrate efflux[1][18]. Cytosolic citrate is then processed into oxaloacetate and acetyl-CoA, which fuels Wakil’s helix for FA elongation. The conversion of oxaloacetate into malate and then pyruvate is producing NADPH, which can be used for the production of NO by iNOS and O2- by NADPH oxidase [31][18]. Indeed, it has been shown that ROS and NO production in LPS-stimulated human macrophages depends on the expression of the citrate efflux transporter SLC25A1[18].
When induced by LPS, citrate efflux from the TCA cycle promotes FA elongation through both acetyl-CoA synthesis and increased levels of NADPH that is an essential cofactor. Produced FA can activate NLRP3, thus promoting inflammation activation. Indeed, fatty acid synthetase (FASN) contributes to NLRP3 activation in LPS-stimulated murine macrophages via AKT activation [36]. When FA synthesis is reduced, IL-1β production is decreased, and the inflammatory response is reduced. In activated macrophages and DCs, FA synthesis is also required for ER/Golgi expansion that supports the secretion of cytokines[37][31][19]. As a result, BMDC maturation triggered by LPS is impaired by FA synthesis inhibition or by silencing of the mitochondrial citrate transporter SLC25A1[1]. In the liver, immunogenic DCs display high content in intracellular lipids[38]. The uptake and storage of FA in lipid droplets is increased when macrophages are activated by TLR4 stimulation due to combined enhanced expression of acyl-CoA synthetase (ACSL1) and acyl-transferases such as diacylglycerol acyltransferase-2 (DGAT2)[39] or glycerol-3-phosphate acyltransferase 3 (GPAT3)[40]. Finally, synthesis of the specific FA arachidonic acid also supports the production of prostaglandins that are important regulators of inflammation[41].
Oxidation of FA in mitochondria produces acetyl-CoA, generating NADH and FADH and fueling the TCA cycle. Upon glucose starvation, FAO induction maintains ATP, NADH, and NADPH levels in monocytes[42]. FAO is also controlled by the energy sensor AMP-activated protein kinase (AMPK)[43], which is activated by low levels of ATP to block anabolic pathways and increase catabolic pathways such as FAO. In M1 macrophages, FAO is low and further suppressed by TLR4 stimulation. Quite similarly, AMPK is downregulated by TLR4 in MoDCs. As a result, FAO is decreased, whereas IL-12 secretion and CD86 membrane expression are enhanced[44]. This functional link between AMPK and FAO activity is supported by observations showing that upon TLR4 stimulation, macrophages and DCs from AMPK-α1 knocked-out mice produce higher levels of IL-6 and TNFα [45]. In contrast, IL-4 stimulates STAT6, thus activating AMPK, resulting in increased FAO in M2 macrophages. Moreover, immunotolerance during the late steps of sepsis is associated with increased levels of FAO[46]. Altogether, this supports the notion that a low level of FAO contributes to inflammation in macrophages and DCs.
This entry is adapted from the peer-reviewed paper 10.3390/immuno1010001
References
- Everts, B.; Amiel, E.; Huang, S.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332.
- Thwe, P.M.; Pelgrom, L.; Cooper, R.; Beauchamp, S.; Reisz, J.A.; D’Alessandro, A.; Everts, B.; Amiel, E. Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses. Cell Metab. 2017, 26, 558–567 e5.
- Curtis, K.D.; Smith, P.R.; Despres, H.W.; Snyder, J.P.; Hogan, T.C.; Rodriguez, P.D.; Amiel, E. Glycogen Metabolism Supports Early Glycolytic Reprogramming and Activation in Dendritic Cells in Response to Both TLR and Syk-Dependent CLR Agonists. Cells 2020, 9, 715.
- Das Gupta, K.; Shakespear, M.R.; Curson, J.E.B.; Murthy, A.M.V.; Iyer, A.; Hodson, M.P.; Ramnath, D.; Tillu, V.A.; von Pein, J.B.; Reid, R.C.; et al. Class IIa Histone Deacetylases Drive Toll-like Receptor-Inducible Glycolysis and Macrophage Inflammatory Responses via Pyruvate Kinase M2. Cell Rep. 2020, 30, 2712-2728.e8.
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and py-ruvate dehydrogenase. Nat. Commun. 2020, 11, 698.
- Everts, B.; Amiel, E.; van der Windt, G.J.W.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012, 120, 1422–1431.
- Schroder, K.; Irvine, K.M.; Taylor, M.S.; Bokil, N.J.; Le Cao, K.-A.; Masterman, K.-A.; Labzin, L.I.; Semple, C.A.; Kapetanovic, R.; Fairbairn, L.; et al. Conservation and divergence in Toll-like receptor 4-regulated gene expression in primary human versus mouse macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E944–E953.
- Liu, T.F.; Vachharajani, V.T.; Yoza, B.K.; McCall, C.E. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 2012, 287, 25758–25769.
- Corcoran, S.E.; O’Neill, L.A. HIF1alpha and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707.
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242.
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826.
- Artyomov, M.N.; Sergushichev, A.; Schilling, J.D. Integrating immunometabolism and macrophage diversity. Semin. Immu-nol. 2016, 28, 417–424.
- Ibrahim, J.; Nguyen, A.H.; Rehman, A.; Ochi, A.; Jamal, M.; Graffeo, C.S.; Henning, J.R.; Zambirinis, C.P.; Fallon, N.C.; Ba-rilla, R.; et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 2012, 143, 1061–1072.
- Amiel, E.; Everts, B.; Fritz, D.; Beauchamp, S.; Ge, B.; Pearce, E.L.; Pearce, E.J. Mechanistic Target of Rapamycin Inhibition Extends Cellular Lifespan in Dendritic Cells by Preserving Mitochondrial Function. J. Immunol. 2014, 193, 2821–2830.
- Moon, J.-S.; Hisata, S.; Park, M.-A.; DeNicola, G.M.; Ryter, S.W.; Nakahira, K.; Choi, A.M.K. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015, 12, 102–115.
- Lawless, S.J.; Kedia-Mehta, N.; Walls, J.F.; McGarrigle, R.; Convery, O.; Sinclair, L.V.; Navarro, M.N.; Murray, J.; Finlay, D.K. Glucose represses dendritic cell-induced T cell responses. Nat. Commun. 2017, 8, 15620.
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430.
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochon-drial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436.
- O’Neill, L.A. A critical role for citrate metabolism in LPS signalling. Biochem. J. 2011, 438, e5–e6.
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614.
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80.
- Perrin-Cocon, L.; Aublin-Gex, A.; Diaz, O.; Ramiere, C.; Peri, F.; Andre, P.; Lotteau, V. Toll-like Receptor 4-Induced Glyco-lytic Burst in Human Monocyte-Derived Dendritic Cells Results from p38-Dependent Stabilization of HIF-1alpha and In-creased Hexokinase II Expression. J. Immunol. 2018, 201, 1510–1521.
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starva-tion-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533.
- O’Neill, L.A. A broken krebs cycle in macrophages. Immunity 2015, 42, 393–394.
- Seim, G.L.; Britt, E.C.; John, S.V.; Yeo, F.J.; Johnson, A.R.; Eisenstein, R.S.; Pagliarini, D.J.; Fan, J. Two-stage metabolic re-modelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat. Metab. 2019, 1, 731–742.
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Suc-cinate Levels. J. Biol. Chem. 2016, 291, 14274–14284.
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.-H.; et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 2018, 556, 501–504.
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938.
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784.
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269.
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662.
- van Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L.; Bekkenkamp-Grovenstein, M.; Hijmans, A.; Engelke, U.F.H.; Wevers, R.A.; et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesi-ty-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313.
- Keiran, N.; Ceperuelo-Mallafre, V.; Calvo, E.; Hernandez-Alvarez, M.I.; Ejarque, M.; Nunez-Roa, C.; Horrillo, D.; May-mo-Masip, E.; Rodriguez, M.M.; Fradera, R.; et al. SUCNR1 controls an anti-inflammatory program in macrophages to reg-ulate the metabolic response to obesity. Nat. Immunol. 2019, 20, 581–592.
- Moon, J.S.; Lee, S.; Park, M.A.; Siempos, I.; Haslip, M.; Lee, P.J.; Yun, M.; Kim, C.K.; Howrylak, J.; Ryter, S.W.; et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J. Clin. Investig. 2015, 125, 665–680.
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23.
- Ibrahim, J.; Nguyen, A.H.; Rehman, A.; Ochi, A.; Jamal, M.; Graffeo, C.S.; Henning, J.R.; Zambirinis, C.P.; Fallon, N.C.; Barilla, R.; et al. Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 2012, 143, 1061–1072.
- Huang, Y.L.; Morales-Rosado, J.; Ray, J.; Myers, T.G.; Kho, T.; Lu, M.; Munford, R.S. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J. Biol. Chem. 2014, 289, 3001–3012.
- Feingold, K.R.; Shigenaga, J.K.; Kazemi, M.R.; McDonald, C.M.; Patzek, S.M.; Cross, A.S.; Moser, A.; Grunfeld, C. Mecha-nisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 2012, 92, 829–839.
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; Di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436.
- Raulien, N.; Friedrich, K.; Strobel, S.; Rubner, S.; Baumann, S.; von Bergen, M.; Korner, A.; Krueger, M.; Rossol, M.; Wagner, U. Fatty Acid Oxidation Compensates for Lipopolysaccharide-Induced Warburg Effect in Glucose-Deprived Monocytes. Front. Immunol. 2017, 8, 609.
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498.
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749.
- Carroll, K.C.; Viollet, B.; Suttles, J. AMPKalpha1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J. Leukoc. Biol. 2013, 94, 1113–1121.
- Liu, T.F.; Vachharajani, V.; Millet, P.; Bharadwaj, M.S.; Molina, A.J.; McCall, C.E. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J. Biol. Chem. 2015, 290, 396–408.