Metformin (MF), a first-line drug to treat type 2 diabetes mellitus (T2DM), alone and in combination with other drugs, restores the ovarian function in women with polycystic ovary syndrome (PCOS) and improves fetal development, pregnancy outcomes and offspring health in gestational diabetes mellitus (GDM) and T2DM. MF treatment is demonstrated to improve the efficiency of in vitro fertilization and is considered a supplementary drug in assisted reproductive technologies. MF lacks teratogenic effects and has positive health effect in newborns. The entry is focused on use of MF therapy for restoration of female reproductive functions and improvement of pregnancy outcomes in metabolic and endocrine disorders.
- : metformin
- diabetes mellitus
1. Introduction
The signaling pathways of metformin (MF) in cells are still not fully understood, they seem to be dependent on species and cell type, as well as doses and routes of administration, along with metabolic and hormonal status of subjects [1][2][3][4][5][6].
The molecule of MF, a small hydrophilic cation, is transported from the extracellular space to the cytoplasm of the target cell through organic cation transporters-1 and -2 (OCT1, OCT2), multidrug and toxin extrusion transporters (MATE), and ATM (ataxia telangiectasia mutated) transporter, and OCT1 and OCT2 are considered as the main functional units of MF transmembrane transport[7]. The transfer of MF across the placental barrier during pregnancy is largely dependent on the transporter OCT3[8]. The ultimate intracellular target for MF is the 5′-adenosine monophosphate-activated protein kinase (AMPK), the key energy sensor of the cell, although MF does not interact directly with the enzyme[1] [9][10][11]. In pathological conditions, like type 2 diabetes mellitus (T2DM) and metabolic syndrome (MetS), the activity of AMPK is reduced. MF’s action increases the activity of AMPK, and consequently normalizes the energy metabolism of the target cell. The AMPK consists of a catalytic α-subunit and the regulatory β- and γ-subunits that form a functionally active αβγ-heterotrimeric complex, and is widely distributed in all subcellular compartments (cytoplasmic, lysosomal, mitochondrial, and nuclear). AMPK is activated by increasing levels of AMP, a positive allosteric regulator of the enzyme[10][11][12][13]. The interaction of AMP with the adenine nucleotides-binding sites located in the γ subunit leads to stabilization of the αβγ heterotrimeric complex and enables phosphorylation of the α-subunit by liver kinase B1 (LKB1), which leads to the increase in AMPK activity [11][12][14] (Figure 1). Activating phosphorylation of AMPK may be also mediated by Ca2+-calmodulin-dependent protein kinase kinase 2 (CaMKK2)[15][16] and transforming growth factor β activated kinase-1 (TAK1) [17][18][19][20], but LKB1 is most important for AMPK activation[10][13][21][22][23]. Allosteric binding of AMP and ADP to γ-subunit of AMPK increases the ability of LKB1 and CaMKK2 to phosphorylate AMPK α-subunit at the Thr172 [24][25][26]. In the lysosomes, the “non-canonical” pathway of LKB1-mediated AMPK activation is carried out through dissociation of fructose 1,6-bisphosphate from aldolase. At the lysosomal surface, free aldolase promotes the formation of a multiprotein complex, including the vacuolar H+-ATPase and the scaffold protein AXIN, and this complex ensures the effective binding between AMPK and LKB1, thereby activating AMPK[27][28]. A negative regulator of AMPK is the protein phosphatase 2C (PP2C), which dephosphorylates and inactivates the α-subunit of AMPK, causing the dissociation of the αβγ-heterotrimeric complex. Elevated levels of AMP lead to an inhibition of PP2C activity, which allows AMPK to remain stable in the active Thr172-phosphorylated state[28][29].
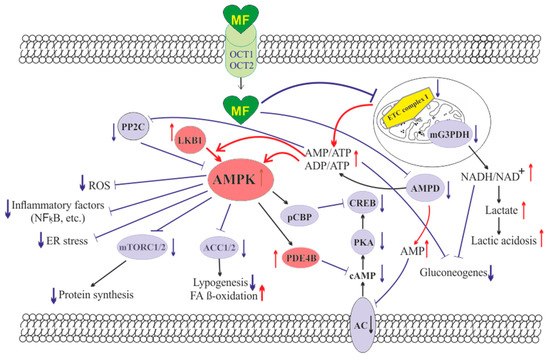
Figure 1. The cellular mechanisms of metformin action which are carried out by activation of the AMP-activated protein kinase and inhibition of the mitochondrial electron transport chain complex I. Abbreviations: AC, adenylyl cyclase; ACC1/2, acetyl-CoA carboxylases 1 and 2; AMPD, AMP deaminase; AMPK, the heterotrimeric AMP-activated protein kinase consisting of the α1/2 (the target for activation phosphorylation at the Thr172), β1/2 and γ1/2/3 subunits; CREB, cAMP-activated transcription factor (cAMP response element-binding protein); ETC complex I, the mitochondrial NADH-dehydrogenase complex, the first complex of the respiratory electron transport chain; FA, fatty acids; LKB1, liver kinase B1; mG3PDH, mitochondrial glycerol-3-phosphate dehydrogenase; mTORC2, the mTOR complex 2; NFκB, nuclear factor κB; OCT1/2, the organic cations transporters 1 and 2; pCBP, the Ser436-phosphorylated form of CREB-binding protein with acetyltransferase activity, a co-activator of the factor CREB; PDE4B, cAMP-specific 3′,5′-cyclic phosphodiesterase 4B; PKA, cAMP-dependent protein kinase; PP2C, protein phosphatase 2C; ROS, reactive oxygen species.
MF penetrates into the mitochondria through intracellular space and accumulates in them. While in the mitochondria, MF inhibits the mitochondrial ETC complex I, which leads to decrease in ATP production and increase in the [AMP]i/[ATP]i and [ADP]i/[ATP]i ratios[30][31][32][33]. Moreover, MF decreases the activity of the enzyme AMP-deaminase (AMPD), which converts AMP to inosine monophosphate, inducing the accumulation of AMP within the cell [34]. The MF-induced increase in the intracellular AMP level leads to the activation of AMPK as described above[21][35]. The MF effect on AMPK activity is observed at drug concentrations below 80 μM, which are achieved with oral administration of therapeutic doses of MF [57]. The MF-induced activation of AMPK results in the stimulation of energy-producing catabolic pathways that mediate the increased glucose uptake by cells, the increased expression and activity of the membrane glucose transporters, the activated metabolic processes such as glycolysis and oxidative phosphorylation, and the normalization of mitochondrial biogenesis[36][3][37][38]. The MF-induced AMPK stimulation leads to phosphorylation of types 1 and 2 acetyl-CoA carboxylases (ACC1 and ACC2), inducing an inhibition of lipogenesis and stimulation of the β-oxidation of free fatty acids [39][40][41] (Figure 1). The ultimate results of this metabolic cascade is the decrease of T2DM- and MetS-produced dyslipidemia, and the normalization of lipid metabolism. In addition, the AMPK activation induces a plethora of cellular events, including regulation of autophagy and apoptotic processes, a decrease in the activity of inflammatory factors, including nuclear factor κB (NF-κB) and interleukin 1β, an inhibition of the ROS production, a decrease in the ER stress, as well as a decrease in insulin/IGF-1-induced activation of the mTORC1/2 complexes and a decrease in the protein synthesis[42][41][43][44][45][46].
The MF is a functional antagonist of cAMP-dependent signaling cascades, which are stimulated by hormones, glucagon in particular, through the Gs protein-coupled receptors and the membrane-bound forms of adenylyl cyclase (AC)[47][48]. The stimulation of AC results in an increase in the intracellular cAMP level and the activation of the protein kinase A (PKA) and the cAMP-activated transcription factor CREB (cAMP response element-binding protein). The MF-induced activation of AMPK promotes phosphorylation and activation of cAMP-specific 3′,5′-cyclic phosphodiesterase 4B (PDE4B), thereby reducing the intracellular level of cAMP[48] Moreover, MF causes an increase in the intracellular level of AMP, a negative regulator of the catalytic site of AC, which leads to inhibition of AC activity and a decrease in cAMP production. An increase in the level of AMP can be the result of both inhibition of the mitochondrial ETC complex I, and suppression of the activity of AMP deaminase [34][49] (Figure 1). A decrease in the activity of cAMP-dependent pathways in the liver, like activation of AMPK, leads to the inhibition of glucose synthesis in hepatocytes. Furthermore, MF-induced AMPK activation induces the protein kinase ι/λ-mediated phosphorylation of cyclic AMP response element binding (CREB)-binding protein (CBP or CREBBP) at the Ser436, which leads to the inability of the phospho-CBP to form a functionally active complex with the factor CREB and thereby inhibits the cAMP-dependent gene transcription[50]
Along with AMPK-dependent, there are also AMPK-independent pathways of MF action on the intracellular effector systems and gene expression. High-dose MF inhibits the activity of the mitochondrial glycerol-3-phosphate dehydrogenase (mG3PDH)[51]. The inhibition of mG3PDH leads to an increase in NADH levels and decreases NAD+ levels, and this causes a deficiency in NAD+, which is involved in the conversion of lactate to pyruvate (Figure 1). Since a decrease in mG3PDH activity inhibits the conversion of lactate to glucose, the result of impaired gluconeogenesis in hepatocytes is an accumulation of lactate, which can cause lactic acidosis in the conditions of high-dose MF treatment[51][52]. Another target of MF is the enzyme H3K27me3-demethylase KDM6A/UTX, which is responsible for the transcriptional activity of a large number of genes[53].
The antidiabetic effects of MF may be due to the changes in the gut microbiota, due to stimulation of the growth of bacteria that produce short-chain fatty acids[54]. By modulating the composition of the microbiota in rodents with T2DM and MetS, MF reduces the levels of bacterial lipopolysaccharides in the blood[55], and activates AMPK-dependent pathways in the mucosal layer of the intestine, reducing glucose absorption[56].
The most important mechanism of action of MF on target cells is the enhancement of the insulin signaling pathways and the decrease in insulin resistance (IR). This may be due to inhibition of hyperactivated nuclear factor κB (NF-κB), a transcription factor that provokes the development of IR, as well as a decrease in the expression of the phosphatase and tensin homolog (PTEN), which dephosphorylates phosphatidylinositol-3,4,5-triphosphate and thereby prevents insulin-induced stimulation of Akt kinase, a key effector component in the 3-phosphoinositide signaling pathway. The inhibitory effect of MF on the activity of NF-κB-dependent signaling pathways is carried out mainly through the stimulation of AMPK[34][57][58]. Since NF-κB plays a key role in inflammatory reactions, its inhibition by MF promotes the weakening of inflammation and increases the cell survival, and these effects of MF are prevented by AMPK inhibitors[34][59][60].
2. Metformin and Polycystic Ovary Syndrome
2.1. Pathophysiology of Polycystic Ovary Syndrome
The polycystic ovary syndrome (PCOS) occurs in average from 9% to 18% of women of reproductive age and includes a number of metabolic and endocrine dysfunctions[61]. Some of them are: (i) the ovarian dysfunction, characterized by irregular or no ovulation (oligo- or amenorrhea), the increased secretion of androgens (hyperandrogenism, HA) and estrogens, the endometrial hyperplasia and the increased size of the ovaries, (ii) the pancreatic dysfunction leading to insulin hypersecretion and, as a result, to insulin resistance (IR) development, (iii) the adrenal dysfunction, which leads to hyperproduction of androgens, and (iv) the functional changes in the hypothalamic and pituitary links of the female hypothalamic-pituitary-gonadal (HPG) axis[62][63][64][65]. Since these dysfunctions and changes are usually associated with obesity, MetS and T2DM, the PCOS is much more common in women with these metabolic disorders (on average in 30% of cases), with a significant proportion of PCOS patients having IR with accompanying compensatory hyperinsulinemia[66][67][68][69][70][71]. According to the Rotterdam criteria (2003), the main diagnostic criteria for PCOS are clinical or biochemical HA, oligo- or amenorrhea associated with chronic anovulation, and morphological features of PCOS, which include 12 or more follicles (2 to 9 mm) in each ovary and/or an increase in ovarian volume over 10 mL[72][73][74]. It should be noted that about 80% of women with anovulatory infertility have typical signs of PCOS[61].
The etiology and clinical manifestations of PCOS depend on many factors, as well as combinations and interactions between them. The genetic predisposition[75][76][77][78] and epigenetic factors, including an increased level of gene methylation, histone modification, and microRNA pattern variation [79][80][81], are important for the development of PCOS. Environmental and socioeconomic factors are also of great importance, including ethnic characteristics, nutrition, and adverse environmental factors (toxins, xenobiotics, chemical mutagens, and ionizing radiation)[82][83][84]., The development of PCOS in women largely depends on the effects of maternal hormones during the prenatal period, as well as on their metabolic and hormonal status in the early childhood[79][85][86][87].
2.2. The Use of Metformin in PCOS Women
In recent years, MF therapy has become widely used for correction of the metabolic and hormonal impairments in women with PCOS and for restoration of their reproductive functions[80][88][89], including the improvement of IVF/ICSI outcomes in PCOS [90][91][92][93]. MF is most effective in treating PCOS patients with the metabolic disorders such as T2DM, obesity, dyslipidemia, and severe IR [94][95][96]. This is majorly attributed to the alleviation of negative effects of these disorders on the female reproduction by MF, increased tissues sensitivity to insulin, improved lipid and glucose metabolism and cell metabolism, and reduced inflammation and oxidative stress in the ovaries as well as in other tissues. In cases where significant metabolic changes in PCOS patients are not observed during treatment, MF therapy can lead to energy and hormonal imbalance. The outcomes may be the opposite of improvement, but a further deterioration in reproductive functions. This possibility is supported by the data from clinical trials on metabolic changes, including an increase in fasting glucose clearance and endogenous glucose production[97][98], as well as changes in the microbiota in non-diabetic individuals[99], as well as data on metabolic and hormonal dysfunctions in normal rodents, for a long time receiving MF[100].
There is a lot of clinical evidence of the high efficacy of MF in PCOS, which makes it feasible to consider MF as a second-line drug for ovulation induction in women with PCOS[101][102][103][104][105][106][107][108]. MF is recommended for the induction of ovulation in PCOS women who are either resistant to clomiphene citrate (CC) or require antiandrogen therapy without the use of contraceptives[105], as well as in PCOS patients with severe obesity and impaired lipid metabolism [109]. One very important consideration during PCOS treatment with MF is that drug has no or little adverse effects on the outcomes of pregnancy as well as the health of fetus and newborn, which indicates the safety of MF therapy[106][107]. The gastrointestinal side effects of MF have been reported in a number of cases, but these effects did not significantly affect the health of PCOS women [92][110].
The MF treatment of PCOS women normalizes the frequency and regularity of ovulation, including when co-administered with exogenous gonadotropins[92][111][112]. This suggests that MF can also affect the sensitivity of ovarian cells to gonadotropins, which is important for the ART. As a result, during the ART, the most promising approach is the combined use of MF with gonadotropins[113][93]. In PCOS, MF improves clinical pregnancy rates and live birth rates[92] [113][92][93][114][115][116][117][118][119], and also reduces the number of miscarriages and increases the rate of embryo implantation[120][121].
There is evidence of a positive effect of MF on the effectiveness of IVF and IVF/ICSI in PCOS women [94]. It is believed to be due to the normalization of metabolic and hormonal parameters and the androgen levels in PCOS, which leads to an improvement of embryo implantation, an increase in the ovarian response to gonadotropins and a decrease in the rates of miscarriage[92][118][122][123][124][125]. The increased gonadotropin sensitivity allows avoiding the use of high-dose gonadotropins and, thereby, preventing the ovarian hyperstimulation syndrome (OHSS), a severe complication of gonadotropin-induced ovulation induction. However, it should be noted that some data on the use of MF in the ART technology in PCOS women are not so unambiguous, and there are results that do not support the efficacy of MF in IVF/ICSI. The clinical studies carried out by Egyptian group of physicians showed no improvement in IVF rates in PCOS women who received MF[126]. However, in this study, overweight or obese PCOS women received short-term courses of low-dose MF (1000 mg/day), from the start of ovarian stimulation with gonadotropins until proof of clinical pregnancy. As a result, in this case, the period of time for the manifestation of the restorative effects of MF on the ovaries and folliculogenesis in PCOS patients may not have been long enough. Potentially, for an adequate estimation of MF effectiveness in PCOS patients it is necessary to separate them in the groups, based on the severity and duration of the disease and in the body mass index [92][94], as well as the severity of IR, dyslipidemia and hyperglycemia.
2.3. Combined Use of Metformin with Clomiphene Citrate, Letrozole, Liraglutide, Saxagliptin, or Oral Contraceptives
A promising approach to treat PCOS is the use of combination of MF with the other drugs that improve the ovarian function and metabolic parameters in PCOS, with the best candidates for co-administration are CC, a mild nonsteroidal estrogen antagonist belonging to the family of selective estrogen receptor modulators, and letrozole, a non-steroidal aromatase inhibitor that prevents the conversion of androgens to estrogens [96][114][127][128][129][130][131][132].
The CC is the main drug of choice for treatment of PCOS, yet a significant proportion of PCOS women have weak or no response to CC therapy. Therefore, a search is underway for drugs that can potentiate the therapeutic effects of CC in PCOS, and MF is one of the most promising candidates [114][122][94][127][128][129][130]. Combined use of MF plus CC in PCOS showed significant improvement in clinical indices of pregnancy and the combination therapy is more effective than the use of CC alone. However, a number of studies reported no effect[133] or relatively weak potentiating effect of MF for CC therapy[117]. One of the possible reasons for these contradictory results may be the difference in the sensitivity to CC and MF in PCOS patients. The most profound potentiating effect of MF on the induction of ovulation and pregnancy rates is found in patients with a pronounced resistance to CC[134][135][136]. However, some PCOS patients may be also insensitive to MF, which is due to many factors, including the polymorphisms and inactivating mutations in the transmembrane proteins facilitating intracellular transport MF[137]. As a result, the combined therapy is expected to benefit mainly PCOS patients with reduced sensitivity to CC, pronounced obesity, IR and dyslipidemia, and high sensitivity to MF.
In recent years, the data have been obtained for the effectiveness of the combined use of MF and letrozole, an aromatase inhibitor that is widely used to restore the ovarian cycle and induction of ovulation and improves oocyte implantation and pregnancy rates in women with PCOS, primarily those with reduced sensitivity to CC[134][132][136]. The combined therapy with MF demonstrated enhancement for the improving effects of letrozole on the pregnancy and live birth rates. Moreover, there are clinical results showing that the combined use of letrozole and MF is more effective than the combined use of CC and MF[129][138].
In PCOS patients, the efficiency of MF therapy is increased when MF is used with oral estrogen-progestin contraceptives, both acting similar, by suppressing ovarian androgen overproduction and normalizing menstrual cycle, most noticeably in obese PCOS women[139][140]. On a contrary, when the same combined treatment (MF and oral contraceptives) is used for PCOS women with normal or reduced body weight, it results in a decrease in their muscles mass, leads to the water retention and the formation of an “osteosarcopenic” phenotype[141]. Two main reasons are behind the decrease in the muscles mass during combined therapy. First, MF and oral contraceptives reduce the blood androgen levels. It is known that in PCOS there is a significant positive correlation between the blood level of androgens and the muscles mass[142]. Second, MF-induced activation of AMPK and changes in mitochondrial energy status stimulate catabolic processes in the muscles tissue, which leads to muscles atrophy, as shown in patients with T2DM [143]. In this regard, it should be noted that MF treatment of T2DM patients leads to an increase in the blood level of fibroblast growth factor 21 (FGF21), which is one of the specific markers of muscles damage and degeneration [144]. Thus, it is highly recommended to take into account the proportion of the muscle tissue and body mass index in PCOS women, as well as the severity of HA when considering the option of using the combined therapy of MF and oral contraceptives[141].
The agonists of glucagon-like peptide-1 (GLP-1) receptor and the inhibitors of dipeptidyl peptidase-4 are widely used to treat T2DM and MetS [145][146][147][148], but they can also be used to correct the metabolic alterations and IR in PCOS women, as well as in pregnant women with GDM and T2DM [149][150]. It is shown that MF enhances the beneficial effect of liraglutide, a selective GLP-1 receptor agonist, on insulin sensitivity and glucose homeostasis. The 12-week treatment of 30 obese PCOS women with a combination of MF (1000 mg twice a day) and liraglutide (1.2 mg/day) causes a decrease in IR and normalizes the sensitivity of patients to glucose, and the combined therapy was more effective than monotherapy[151]. The treatment of premenopausal PCOS women with MF (2000 mg/day), saxagliptin (5 mg/day), an inhibitor of dipeptidyl peptidase-4, or a combination of MF and saxagliptin leads to normalization of glucose tolerance on average of 56% of patients[152]. Moreover, in the group treated with MF alone or saxagliptin alone, the improvement of glycemic control is demonstrated only in 25 and 55% of patients, respectively, while the combined therapy restores glucose tolerance in 91% of women with PCOS[152]. A high efficacy of the combined therapy was shown by other group of authors who monitored the 16-week treatment of 38 women with pre-diabetes and PCOS using the MF plus saxagliptin[153]. Weight loss and decrease in hyperglycemia and IR, which are induced by treatment of obese PCOS patients with GLP-1 receptor agonists, lead to a decrease in HA[154][155][156] and an improvement in menstrual frequency[154][156]. Liraglutide, an analogue of GLP-1, normalizes the menstrual cycle and fertility in women with HAIR-AN syndrome, which is due to a decrease in the levels of androgens and insulin[157]. Consequently, in PCOS patients, MF-induced potentiation of the metabolic-improving effects of GLP-1 agonists may also increase their restorative effects on the menstrual cycle and fertility.
2.4. The Mechanisms of Metformin Effects on Reproductive Functions in PCOS
2.4.1. Metformin-Induced Inhibition of Hyperandrogenism and Normalization of the Steroid Hormones Balance
One of the main mechanisms of the restorative effect of MF on ovarian function, ovulation and pregnancy in PCOS women mediates through the pronounced antiandrogenic effect of MF, both in monotherapy and in combination with other drugs[125][158][159][160][161][162][163][164][165][166][167]. One-year treatment of overweight PCOS women with MF (1700 mg/day) reduced the levels of free testosterone, dehydroepiandrosterone (DHEA) and androstenedione, and significantly weakened the signs of hirsutism. This effect of MF was strongly associated with a decrease in the homeostasis model assessment of insulin resistance index (HOMA-IR) and an improvement in glucose tolerance, but was weakly associated with a decrease in the body weight, which indicates a main contribution of a decrease in IR and hyperinsulinemia to the antiandrogenic effect of MF[168]. An antiandrogenic effect was demonstrated in the treatment of overweight and obese adolescents with MF (1000–2000 mg/day), and was accompanied by a significant decrease in IR [158][161][162][169][170][164][165]. MF reduced both the basal and gonadotropin-stimulated testosterone levels, and these effects were observed even with short-term MF treatment. The administration of MF for two days to PCOS women caused a decrease in their testosterone levels stimulated by luteinizing hormone (LH). This effect was not due to a decrease in the body weight and the changes in metabolic indices, pointing to potential direct influence of MF on steroidogenic activity in ovarian cells[171].
In PCOS, the severity of IR is positively correlated with the severity of HA and dysregulations of the ovulatory cycle. The PCOS women with oligomenorrhea and without HA usually do not have IR, while the PCOS women with oligomenorrhea and HA often show significant signs of IR[172]. In turn, in PCOS women with regular ovulatory cycle, IR was less pronounced than in women with PCOS and irregular or no ovulation[173].
The inhibitory effect of MF on the production of steroid hormones by ovarian cells was demonstrated in the in situ experiments using different cell lines[74][174][175][176]. Cultured human ovarian cells grown in the presence of MF, showed a decrease in production of basal and gonadotropin- or insulin-stimulated steroid hormones. Similar effects were shown for progesterone and estradiol in granulosa cells and androstenedione in theca cells. Inhibitory effect of MF was dose-dependent and most pronounced in the measure of suppression of hormone-stimulated steroidogenesis [175]. MF (10 mM) treatment of bovine granulosa cells isolated from small follicles led to a decrease in both the basal and follicle-stimulating hormone (FSH)- and IGF-1-stimulated production of progesterone and estradiol[176].
When deciphering the mechanisms of the inhibitory effect of MF on steroidogenic activity in the ovaries, a key role is assigned to stimulation of AMPK, and the triggering of AMPK-dependent pathways in ovarian cells [74][176][177][(Figure 2). The mechanisms of AMPK activation in ovarian cells are the same as described in the Section 2 above, and are triggered by MF-induced inhibition of electron transport chain in mitochondrial respiratory complex I[178]. It is worth noticing that in humans, other mammals and birds (cows, goats, sheep, pigs, rats, mice, chicken), AMPK is widely expressed in different types of the ovarian cells (oocyte-cumulus complexes, granulosa cells, and theca cells) and in the corpus luteum[176][177][179].
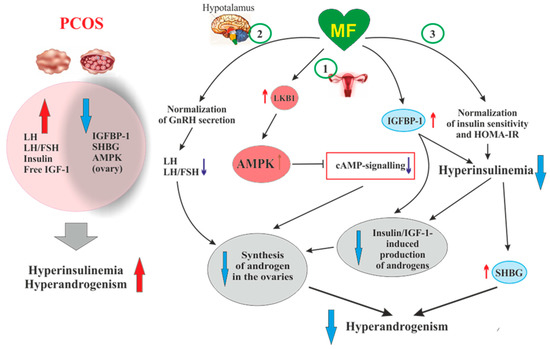
Figure 2. The pathways involved in the inhibitory effect of metformin on hyperandrogenism in PCOS. Hyperinsulinemia and HA are among the key pathogenetic factors in the development of PCOS, which is why, their attenuation by MF is the most important mechanism for improving effect of this drug on ovarian function in PCOS women. In PCOS, MF-induced increase in insulin sensitivity leads to a decrease in the HOMA-IR and a weakening of compensatory hyperinsulinemia. Another mechanism for lowering insulin levels may be an increase in the level of IGFBP-1, which specifically binds insulin and IGF-1. In PCOS, the expression of IGFBP-1 is generally reduced, and MF treatment may be one way to normalize it. A reduced hyperinsulinemia and an increase in IGFB-1 levels lead to a decrease in the stimulating effect of insulin and IGF-1 on the ovarian steroidogenesis and a weakening of HA. Hyperinsulinemia leads to a decrease in the production of SHBG, which provokes HA in PCOS. MF-induced reduction of hyperinsulinemia leads to the normalization of the SHBG levels, thereby preventing excess androgen levels in the blood. By improving the functionality of the hypothalamic signaling network responsible for the pulsatile secretion of GnRH, treatment with MF leads to the normalization of blood LH levels and the LH/FSH ratio, both of which are increased in PCOS. A decrease in blood LH levels results in a weakening of gonadotropin-induced androgen production by the ovaries. A direct regulatory effect of MF on ovarian steroidogenesis was also established. By inhibiting the mitochondrial ETC complex I, stimulating the LKB1 activity and, as a result, increasing the AMPK activity, MF reduces the synthesis of androstenedione in the ovarian cells and prevents HA. It can be assumed that the prevalence of some mechanisms of the inhibitory effect of MF on HA is due to the characteristic features of PCOS pathogenesis and the metabolic and hormonal status of the ovaries. Details and bibliographic references are presented in the Section 2.4. Abbreviations: AMPK, AMP-activated protein kinase; FSH, follicle-stimulating hormone; HA, hyperandrogenism; HOMA-IR, homeostasis model assessment of insulin resistance; IGF-1, insulin-like growth factor-1; IGFBP-1, insulin-like growth factor-binding protein-1; LH, luteinizing hormone; LKB1, liver kinase B1; SHBG, androgen and sex hormone-binding globulin.
There is a large body of experimental data that AMPK is essential for the regulation of folliculogenesis and meiotic activity, both control the maturation of oocytes[176][180][181][182][183][184], and that AMPK is involved in the regulation of steroidogenesis in ovarian granulosa cells[177][185]. Deletion of the AMPK α1-subunit in mouse oocytes leads to a 27% decrease in litter size, and after IVF, the number of embryos in these mutant mice decreases by 68%[186]. In the ovaries of mutant mice, the levels of transmembrane connexin-37 and N-cadherin, which mediate the intercellular communication and are involved in the formation of the oocyte-cumulus complexes, were significantly reduced. The activity level within cAMP-dependent cascade, which includes PKA and factor CREB, and the activity of mitogen-activated protein kinase (MAPK) cascade are reduced, indicating weakening of cAMP- and MAPK-mediated signal transduction[185][186]. The components of these signaling pathways are involved in the junctional communication between the oocyte and the cumulus/granulosa cells. The MII oocytes in mice lacking the α1-AMPK have a significantly reduced intracellular ATP level and decreased levels of cytochrome c and peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α), which indicates the impaired mitochondrial biogenesis and the activation of apoptotic processes [186].
In ovarian cells, through the activation of AMPK, MF inhibits the cAMP signaling pathways, decreases the expression and activity of the steroidogenic enzymes and the production of androstenedione, a precursor of testosterone (Figure 2). When exposed to cultured human theca cells, both in the basal and forskolin-stimulated state, application of MF (50 and 200 μM) caused the AMPK stimulation and reduced androstenedione synthesis in dose-dependent manner. In theca cells stimulated by forskolin, a non-hormonal AC activator, MF suppressed the expression of StAR and Cyp171a genes encoding StAR protein, which carries out cholesterol transport into mitochondria (the first, rate-limiting stage of steroidogenesis), and cytochrome P450c17α, which catalyzes the synthesis of androstenedione[172]. The inhibitory effect of MF on steroidogenesis in the rat and bovine granulosa cells was also due to AMPK activation, as indicated by an increase of Thr172 phosphorylation of AMPK α-subunit, as well as an increase of Ser79 phosphorylation and inhibition of the main target of AMPK, the enzyme acetyl-CoA-carboxylase[176][187]. The involvement of AMPK in the antiandrogenic action of MF is supported by the data on a similar effect of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), the pharmacological activator of AMPK[176]. The MF-induced stimulation of AMPK in granulosa cells results in a decrease in the activity of MAPK cascade, primarily the kinases ERK1/2, and inhibition of phosphorylation of MAPK-activated protein kinase-1, also referred to as ribosomal s6 kinase (p90RSK)[176][187][188]. Therefore, through the AMPK-dependent mechanisms, MF not only reduces the excessive steroidogenic activity in ovarian cells, but also suppresses and/or modulates the cell growth and intracellular protein synthesis.
An important role of AMPK-dependent mechanisms in the antiandrogenic action of MF in PCOS is supported by the data on the relationship between the androgen production and the activity of LKB1 in the ovaries of mice with experimental HA[189][190][191][192][193] . The LKB1 expression in the ovaries of hyperandrogenic mice is inhibited by high concentrations of androgens through activation of intracellular androgen receptors. In opposite, LKB1 activation leads to a decrease in the androgens production by theca cells, but increases the estrogen production by granulosa cells. Transgenic mice overexpressing LKB1 are characterized by the increased resistance to the development of HA[189]. Mice with a functionally inactive ovarian gene Lkb1 have significantly enlarged ovaries and activated entire pool of primordial follicles, but without further maturation and ovulation, which results in the premature ovarian failure and severely reduced fertility[194]. The data indicates that MF-induced activation of LKB1/AMPK pathway in PCOS ovaries normalizes ovarian steroidogenesis and counteracts HA (Figure 2).
Another, very important mechanism of antiandrogenic action of MF is largely due to an MF-induced increase in insulin sensitivity and consequent weakening of compensatory hyperinsulinemia, the main pathogenic factor in PCOS, also closely associated with HA[195][196][197] (Figure 2).
It is generally accepted that the stimulating effect of hyperinsulinemia on the production of androgens by ovarian cells is based on low affinity binding of insulin with IGF-1 receptors[197][198][199]. In the 1990s, it was shown that insulin in vitro and in vivo activates the IGF-1 receptor in the ovaries, which leads to an increase in the synthesis and secretion of androgens. This is supported by the data on the increased secretion of androstenedione and the elevated basal and LH-stimulated production of testosterone in cultured ovarian theca and stroma cells incubated in the presence of insulin[198][200]. A decrease in insulin secretion induced by MF (500 mg three times daily) in obese PCOS women led to inhibition of cytochrome P450c17α activity in the ovaries, decreasing the basal levels of 17α-hydroxyprogesterone and the levels of this hormone stimulated by leuprolide, a gonadotropin-releasing hormone (GnRH) analogue[159].
Along with the activation of IGF-1 receptor, hyperinsulinemia reduces the production of insulin-like growth factor-binding protein-1 (IGFBP-1) by ovarian granulosa cells[198] Figure 2). This protein specifically binds IGF-1, decreasing the concentration of free IGF-1 and weakening its stimulating effect on the IGF-1 receptor and steroidogenesis in the ovarian theca and stromal cells. The result of insulin-induced decrease in IGFBP-1 level is overproduction of androgens and the impaired folliculogenesis and ovulatory cycle[201]. It should be noted that IGF-1, like insulin, reduces the IGFBP-1 production in ovarian cells, while FSH stimulates the IGFBP-1 production, preventing IGF-1-induced stimulation of androgen production [201]. The blood IGFBP-1 levels in PCOS women are significantly lower compare to than in healthy women, which may indicate suppression of IGFBP-1 production under the conditions of hyperinsulinemia[195][197]. At the same time, there is no strong correlation between the IGFBP-1 deficiency and the severity of hyperinsulinemia and IR, which suggests the presence of additional mechanisms mediating the inhibition of IGFBP-1 production in PCOS[197]. There are no data for the MF effect on blood IGFBP-1 levels in PCOS, but there is evidence of its significant increase in MF-treated women with GDM[190] [190]. MF caused an increase in both phosphorylated and non-phosphorylated forms of IGFBP-1, thereby reducing the negative effect of IGFBP-1 deficiency on the course and outcomes of pregnancy[190].
In PCOS, the blood levels of androgen and sex hormone-binding globulin (SHBG) are reduced, which leads to an increase in free testosterone level and free androgen index. As early as the 1990s, hyperinsulinemia was found to be an important factor for the suppression of SHBG production in PCOS[153][191][192]. Overweight plays a key role in this process, as supported by observation that weight loss in obese PCOS women induced by a low-calorie diet leads to a restoration of SHBG levels, which may be due to a decrease in IR and insulin levels[193]. MF administration increases the production of SHBG, by reducing body weight and hyperinsulinemia, and, thereby, reduces the signs of HA in PCOS women[153][202][203] (Figure 2). Blood SHBG levels are also increased in MF-treated obese women without clear signs of PCOS [204]. The PCOS women with low SHBG levels are more sensitive to MF therapy, while the effectiveness of MF in patients with normal or high SHBG levels and, as a consequence, without signs of HA is significantly less noticeable. The PCOS women with an average SHBG level in the blood of 37.5 nmol/L respond well to MF treatment, while in PCOS women with an average SHBG level of 56.0 nmol/L, the response to MF was weak[205]. Therefore, the assessment of the blood SHBG concentration is an important prognostic factor for predicting the effectiveness of MF therapy in PCOS. Thus, a MF-induced decrease in the level of insulin should lead to a weakening of the stimulating effect of insulin on the ovarian IGF-1 receptors, prevent their stimulation by an excess of free IGF-1, and reduce the blood level of free androgens by restoring the SHBG production.
Of great importance for normalization of the steroidogenic function in the ovaries can be MF-induced decrease in the blood LH level and the LH/FSH ratio, which are significantly increased in PCOS[206][207][208][209] (Figure 2). An increase in the LH/FSH ratio due to abnormal gonadotropin pulsatility and hypersecretion of LH by the pituitary is a significant factor responsible for the deterioration of folliculogenesis and oogenesis in PCOS [209][210][211][212][213][214]. In most cases, gonadotropin imbalance is found in PCOS women with obesity, and level of increase in LH is correlated with the severity of obesity [211]. The restoration of this ratio leads to normalization of the ovulatory cycle and triggers the development of the dominant follicle, improving the rate and outcomesof pregnancy[213][215]. Eight-week treatment of PCOS women with MF (1500 mg/daily) results in a 32% decrease in the blood LH levels and a 42% decrease in the LH/FSH ratio[208]. There is reason to believe that, as in the case of SHBG, the sensitivity of PCOS women to MF therapy depends on the LH level and the LH/FSH ratio. The MF treatment of PCOS women with severely impaired gonadotropin secretion and significantly increased LH level is more effective than the same treatment of PCOS women without gonadotropin imbalance[216].
It is suggested that the restoration of normal LH secretion by the pituitary gland may be due to MF-induced normalization of AMPK-dependent signaling in hypothalamic neurons secreting GnRH[217] (Figure 2). This is due to the ability of MF to cross the blood-brain barrier and reach the hypothalamus and the other brain regions[218] The secretion of GnRH is under the control of neuropeptides, such as kisspeptin, melanocortins, agouti-related peptide and neuropeptide Y, as well as γ-aminobutyric acid and the other biogenic amines[219], and the neurons producing these neurohormones may also be pharmacological targets for MF. The restoration of functional interaction between GnRH-expressing neurons and the other components of the neuronal network responsible for hypothalamic control of the HPG axis can prevent PCOS-associated HA and provide a balance between steroid hormones, thereby normalizing the functionality of feedback loops in this axis. Our group and other authors have demonstrated the restoring effect of MF on leptin signaling pathways in the hypothalamus of animals with metabolic disorders and IR [220][221][222][223], and the improvement of hypothalamic leptin signaling can also make a significant contribution to the restoration the reproductive functions in PCOS. It should be noted that MF, both at the periphery and in the CNS, acts on the signaling and effector systems synergistically with leptin. It is generally accepted that leptin is the most important regulator of the female and male reproductive systems, which stimulates the activity of hypothalamic GnRH-expressing neurons, and affects the other links of the HPG axis[224][225].
2.4.2. Protective Effect of Metformin against Excess Androgens in PCOS
In addition to reducing HA in PCOS, MF is able to prevent the negative effect of excess androgens on the ovarian cells[226][227]. It was shown in mice model of PCOS, that treatment using MF (500 mg/kg, 20 days) after DHEA induction improved the quality of oocytes and normalized the early stages of embryonic development. In the ovaries of MF-treated mice, the restoration of the number of metaphase II oocytes, mitochondrial membrane potential and ATP levels were shown. Along with this, MF attenuated oxidative stress, as indicated by a decrease in reactive oxygen species levels and an increase in the reduced form of glutathione[226].
The inhibition of endoplasmic reticulum (ER) stress and the prevention of MAPK cascade hyperactivation make a significant contribution to protective effects of MF in PCOS-associated HA. The activation of ER stress in the ovaries and the triggering of signaling pathways induced by the unfolded protein response lead to impaired synthesis and post-translational modification of proteins and the mitochondrial dysfunction, all of which negatively affects folliculogenesis and meiotic maturation of oocytes[228][229][230]. The effector components of MAPK cascade p38-MAPK is important for the activation of the unfolded protein response signaling and apoptosis in ovarian cells[231][232]. More recently, it was shown that an excess of androgens led to activation of ER stress and apoptosis in human and mouse cumulus cells[227][233]. The MF treatment reduces ER stress and inhibits p38-MAPK phosphorylation, which is significantly increased in cumulus cells of PCOS women and in the granulosa cells and the oocyte-cumulus complexes in mice with DHEA-induced PCOS [237]. There is every reason to believe that this effect of MF is based on its ability to inhibit HA.
2.4.3. Effects of Metformin on FSH-Activated Signaling in the PCOS Ovaries
Another mechanism of the MF restoring effect on ovarian function in PCOS is the inhibition of expression of the Cyp19a1 gene encoding aromatase. Reduced levels of aromatase result in a decrease in estrogen response to FSH, insulin and IGF-1 in the ovaries[234][235][236]. A large number of PCOS patients have increased sensitivity of granulosa cells to stimulation with FSH, insulin, or IGF-1. This is due to the fact that in granulosa cells of PCOS women, the expression of the FSH and IGF-1 receptors and the IRS1 and IRS2 proteins are significantly increased[237][238][239][240][241]. In addition, the expression of PTEN, a negative regulator of signaling pathway involving insulin/IGF-1 receptors, IRS proteins, phosphatidylinositol 3-kinase (PI 3-K) and Akt-kinase, is reduced, which leads to hyperactivation of Akt-kinase by insulin and IGF-1[241]. In the PCOS ovaries, the important mechanism of suppression of PTEN expression and hyperactivation of the insulin and IGF-1 signaling pathways is an increase in the expression of two microRNAs of miR-200 family, miR-200b and miR-200c, which negatively affect the expression of the PTEN gene[241] In addition, a decrease in the expression of miR-99a, a negative regulator of IGF-1 receptor expression, leads to an increase in the sensitivity of granulosa cells to IGF-1. An increase in the expression and activity of the receptor and postreceptor components of the FSH-, insulin- and IGF-1-regulated signaling systems in PCOS results in the accelerated growth and proliferation of the ovarian cells, primarily granulosa cells, in the response to the stimulating effect of these hormones. Moreover, this potentiates already pre-existing increased ovarian reactivity and premature luteinization[242][243].
MF reduces the expression of FSH receptors thereby weakening the stimulating effects of FSH on steroidogenesis and proliferation of granulosa cells, increased in PCOS, which leads to the normalization of folliculogenesis and ovulation. Under the conditions of ovarian dysfunctions in PCOS, MF treatment postpones the triggering of processes that ensure the normal growth of antral follicles, thus providing more appropriate window of time required for their differentiation and development (on average about three months)[235]. By reducing the ovarian sensitivity to FSH, MF prevents the OHSS, the most common complication of gonadotropin-stimulated induction of ovulation [244][245][135,136,138,255,256].
The inhibitory and modulating effects of MF on the effector components of gonadotropin-stimulated cascades in ovarian cells can be realized through both AMPK-dependent and AMPK-independent pathways, including the MAPK cascade [244,245]. Through AMPK-independent pathways, MF reduces FSH-induced increases in aromatase activity and estradiol synthesis in granulosa cells, and this effect is not reproduced when using AICAR [245]. The inhibitory effect of MF on the expression and activity of aromatase can be elicited through at least three well understood mechanisms.
The first mechanism is MF-induced inhibition of the expression of FSH receptor in granulosa cells, which reduces the stimulatory effect of FSH on the intracellular signaling pathways through which FSH controls the expression of aromatase and steroidogenic enzymes [245]. As noted above, in granulosa cells of women with PCOS, the expression of the Fshr gene is often significantly increased, which causes the elevated responsiveness of the ovaries to FSH [247,248,249]. The polymorphisms in the Fshr gene can have a significant role in modulating the responsiveness to FSH in both, positive and negative way, although in PCOS the data on the interrelation between Fshr isoforms and the activity of FSH receptor are contradictory [257]. At the same time, there is evidence that some polymorphisms can lead to an increase in the sensitivity of FSH receptor to gonadotropin [258,259]. The second mechanism of the inhibitory effect of MF on aromatase activity is due to a decrease in FSH-induced phosphorylation of the transcription factor CREB, which positively regulates the expression of the aromatase gene, as well as the Star, CYP11a1 and HSD3b genes encoding the cholesterol-transporting protein StAR, cytochrome P450scc (CYP11A1), which catalyzes the synthesis of pregnenolone, and 3β-hydroxysteroid dehydrogenase (3β-HSD), which converts pregnenolone to progesterone [245]. The third mechanism involves inhibition of FSH-induced dephosphorylation of the CREB-regulated transcription coactivator 2 (CRTC2) and its translocation into the nucleus, where CRTC2 is involved in the assembly of CREB containing activating transcriptional complex [245]. Thus, MF inhibits the formation of the CREB-CBP-CRTC2 activation complex, which is capable of binding to the CRE regulatory elements in the promoter of the genes encoding aromatase and some steroidogenic proteins, and prevents their overexpression by FSH.
The FSH- and insulin/IGF-1-activated signaling pathways in ovarian cells are closely interrelated due to cross-talk between them, including their interaction through the PKA/PI 3-K/Akt pathway [260,261]. The functional activity of Akt kinase is increased as a result of these pathways activation in the conditions of overstimulation of FSH receptor and FSH-mediated activation of IRS1 protein. This leads to an increase in the survival of ovarian cells, suppresses atresia of the follicles and impairs maturation of the dominant follicle.
The FSH-dependent pathways are also regulated by the members of the transforming growth factor β (TGF-β) family, including activins, inhibins, and anti-Müllerian hormone (AMH). Activins increase the FSH receptor expression and enhance the stimulatory effects of FSH on ovarian steroidogenesis [262,263]. In contrast, AMH and inhibins suppress the stimulatory effects of FSH on folliculogenesis [249,262,264,265]. The regulatory effects of the protein members of the TGF-β-family on the FSH-dependent signaling pathways can be realized through both Akt- and cAMP-dependent mechanisms [266,267,268,269], but the effect of MF on the TGF-β-mediated regulation of the FSH signaling pathways in granulosa cells remains poorly understood.
2.4.4. The Effect of Metformin on the Production of Anti-Müllerian Hormone in PCOS
In PCOS, one of the targets of MF therapy is AMH, a dimeric glycoprotein that is produced by the granulosa cells of the primary, preantral and small antral follicles [270,271]. AMH concentration in the blood of women positively correlates with the follicular reserve and, as a result, in PCOS, the blood levels of AMH are usually increased by two or more fold [251,271,272,273,274]. Excess levels of AMH lead to an impaired folliculogenesis, preventing the recruitment of primordial follicles into the pool of growing follicles and reducing the responsiveness of growing follicles to FSH [249,275,276,277]. The increased levels of AMH may be due to HA and hyperinsulinemia, which are characteristic features of PCOS and are closely interrelated [249,273,278,279,280], as well as to an increase in blood LH levels or the sensitivity of granulosa cells to LH, typical for PCOS patients [280,281,282,283]. In in vitro experiments using lutein granulosa cells obtained from oligo/anovulatory PCOS women, the LH increases the AMH production, while the expression of type II AMH receptors in these cells does not change significantly. In the case of lutein granulosa cells obtained from healthy women and normo-ovulatory PCOS women, the stimulating effect of LH on AMH production is almost completely inhibited, but the inhibiting effect of LH on the expression of type II AMH receptors is preserved. This effect was reproduced with the use of cAMP analogs, which indicates the participation of cAMP-dependent mechanisms in it [281]. All this indicates that PCOS women with an impaired ovulatory cycle have an increase in both the LH-induced AMH production and the responsiveness of lutein granulosa cells to this factor.
By lowering insulin and androgen levels and normalizing gonadotropin levels, MF attenuates ovarian AMH secretion, which leads to a decrease of its inhibitory effect on folliculogenesis and a weakening of the signs of PCOS [270,271,284,285,286,287,288]. Eight-week treatment of PCOS women with MF (1500 mg/day) reduced the blood AMH levels from 10 ± 3.75 to 7.8 ± 3.7 ng/mL [271]. Six-month treatment with MF at the same dose led to a decrease in AMH, ovarian volume and antral follicle number in PCOS women [270]. The treatment of PCOS women with MF at the doses of 850 mg/day (first week), 850 mg/12 h (second week) and 850 mg/8 h (next six weeks), along with the restoration of ovulation and the normalization of LH and testosterone levels, caused a decrease in the blood AMH levels, from 8.99 ± 0.99 to 6.28 ± 0.46 ng/mL [286]. The combined therapy with MF and resveratrol of rats with DHEA-induced PCOS reduced ovarian size, improved ovarian follicular cell architecture, and decreased AMH production [217]. It is assumed that in PCOS, a decrease in the blood AMH level to control values can be considered as one of the prognostic factors of the effectiveness of MF therapy [285,286,289].
However, there are clinical studies that showed a weak suppressive effect of MF therapy on AMH production in PCOS [290], or the absence of this effect [291,292]. This may be due to differences in the MF doses, the duration of MF treatment, and the peculiarities of the hormonal status in PCOS patients. MF treatment of PCOS women for 8 months led to a decrease in the blood AMH level, while four-month MF therapy had a little effect on AMH concentration, although it significantly reduced the blood level of androstenedione and normalized the regularity of the menstrual cycle [284]. It is possible, that normalization of the AMH level at the first stage requires normalization of androgens, insulin and gonadotropins levels, which in turn affects the expression and secretion of AMH. On the other hand, Iraqi scientists showed that the treatment of PCOS women with MF (500 mg three times daily) for three or six months significantly reduced the blood AMH levels, but in the case of six-month therapy, the MF effect became less pronounced [289]. There is reason to believe that the severity of obesity, as well as the degree of an increase in AMH levels, can have a significant effect on the inhibitory effect of MF on ovarian AMH production. The most pronounced inhibitory effect of MF on AMH production was demonstrated in PCOS women with a higher body mass index, as well as with a higher level of AMH [285].
Based on the above results, as well as on the available data on the molecular mechanisms mediating the regulation of AMH production by granulosa cells [249,293], it can be assumed that the main mechanism for the improving effect of MF on AMH levels in PCOS is the weakening of HA. Under normal conditions, the androgens produced by theca cells induce a decrease in AMH levels, which leads to inhibition of the antral follicle development and precedes ovulation. With prolonged exposure to high concentrations of androgens, which are comparable to those in PCOS, the response of granulosa cells to androgens is impaired, resulting in the absence of an androgen-induced fall in AMH levels and dysregulation of follicular development [293]. When PCOS patients are treated with MF, their androgen levels are normalized, and hyperinsulinemia, which is usually associated with HA, is reduced, which leads to the restoration of the granulosa cell response to androgens. Accordingly, the low efficacy of MF in reducing AMH levels and restoring follicular maturation in patients with PCOS may be due to initially mild HA and IR. It is impossible to exclude the direct effects of MF on the production of AMH by follicular cells, including through AMPK-dependent mechanisms, as well as through weakening the stress of the endoplasmic reticulum, stimulated by high concentrations of androgens [237]. However, this issue has not yet been studied.
There are only two clinical studies on the MF effect on the levels of other TGF-β family factors. One of them showed the normalization of the TGF-β level in the blood of PCOS women with MF and cyproterone acetate/ethinyloestradiol treatment [294]. The other authors demonstrated that a long-term therapy with MF (more than three months) led to a significant decrease in the blood level of inhibin-B in PCOS patients [289].
2.4.5. Effect of Metformin on Metalloproteinases in PCOS
Women with PCOS usually have the increased serum levels of type 9 matrix metalloproteinase (MMP-9) and MMP-2 [295,296], and the altered concentrations of the types 1 and 2 tissue inhibitor of MMP (TIMP-1 and TIMP-2) [295]. The changes in the concentrations and balance of MMPs and TIMPs lead to the remodeling in the ovarian stroma, increased ovarian angiogenesis and impaired folliculogenesis [295,296]. The activation of AMPK causes a decrease in the activity of the pathway involving mTOR (mammalian target of rapamycin) and ribosomal protein kinase p70S6K, and suppresses the expression and functional activity of MMPs. Consistently, stimulation of Akt kinase and mTOR enhances the expression of MMPs and, thereby, stimulates angiogenesis, cell migration, cell proliferation and protein synthesis [297,298].
By activating AMPK, MF reduces the expression and activity of MMPs, and this is largely responsible for the well-described antitumor effect of MF [299]. MF treatment (10 mM) of cultured human ovarian granulosa (HTOG) cells, leads to an increase in AMPK activity, an inhibition of mTOR-dependent cascade, and a decrease in the expression of MMP-2 and MMP-9 [300]. Another MF-mediated mechanism for down regulating the MMP expression involves triggering the H19/miR-29b-3p signaling pathway. MF increases the expression of miR-29b-3p, a negative regulator of the MMP-2 and MMP-9 expression via decreasing the expression of histone H19 through methylation of the promoter in the gene encoding this histone, which leads to a decrease in the production of these enzymes and contributes to normalization of ovarian morphology in PCOS [300]. MF-induced activation of the H19/miR-29b-3p pathway is attenuated by the increased expression of histone H19, which decreases the expression of miR-29b-3p. The miR-29b-3p-mediated effect of MF on MMP expression is independent of AMPK, in contrast to MF regulation of mTOR signaling cascade [300].
2.4.6. Influence of Metformin on Inflammation and Lipid Status in PCOS
Dyslipidemia, oxidative stress and inflammation, along with IR, are essential for the pathogenesis of PCOS. These factors are similar to those in MetS and T2DM [301,302]. As a result, the ability of MF to improve energy metabolism, prevent lipotoxicity and restore the redox balance in the ovaries is another mechanism of its beneficial effect on folliculogenesis and the ovulatory cycle in PCOS. In human granulosa cells, through AMPK-dependent mechanisms, MF suppresses the tumor necrosis factor-α (TNF-α)-induced production of pro-inflammatory cytokines, interleukin-8 and chemokine CXCL1/GROα [303]. The other AMPK activators, such as AICAR and Baicalin, also reduce TNF-α- and chemokine-mediated inflammatory responses in ovarian cells [303,304]. In PCOS ovarian cells, the mechanism of MF suppressive effect on the TNF-α-induced cascades includes a decrease in the gene expression of TNF-α [217].
The severity of metabolic disorders positively correlates with the therapeutic effect of MF, which is due not only to its effect on the ovaries, but also to the effect on the metabolic processes in the other tissues. A prominent feature of PCOS is decreased level of high-density lipoprotein cholesterol (HDL-C), and MF is most effective in PCOS patients with blood HDL-C levels below 50 mg/dL [226]. HDL-C modulates glucose homeostasis through AMPK-dependent mechanisms, whereby in PCOS, MF-induced AMPK activation may significantly contribute to the insulin sensitivity-restoring effect of MF [226].
2.5. The Sensitivity of PCOS Women to Metformin Therapy
The responsiveness of PCOS women to MF may be due to the efficiency of MF transport into the target cell and the distribution and pharmacokinetics of this drug in the tissues [28] (Figure 3). Despite the ability to freely penetrate the blood-tissue barriers and redistribute in the tissues, MF transport into the cell is carried out through the specialized transporters, including OCT1, OCT2, ATM and MATE. A decrease in their functional activity due to inactivating mutations in their genes, primarily in the OCT1 gene, has a negative effect on the therapeutic effect of MF, in some cases making PCOS patients completely resistant to MF [151,305,306]. The incidence of OCT1, OCT2 and ATM polymorphisms in PCOS is much higher than in women without PCOS. During genotyping of PCOS patients, non-functional alleles of the OCT1, OCT2, and ATM genes were detected in 29.8%, while low-functional alleles in 57.9% of cases. Non-functional alleles in the OCT1 and OCT2 genes were associated with poor response to MF, as well as with glucose intolerance and significantly elevated levels of proinsulin C-peptide after glucose loading [306]. In the recent work of Taiwanese scientists, the polymorphisms rs683369 (allele G) and rs628031 (allele A) were identified in the OCT1 gene of PCOS women, and they were associated with a decrease in sensitivity of patients to both the MF and insulin therapy [151]. It should be noted that of the 38 currently identified OCT1 gene polymorphisms, the Met408Val (rs628031) variant exhibits the most suppressed MF therapeutic effect [307]. Quite unexpected finding was that single-nucleotide polymorphism in the OCT2 gene did not significantly affect the affinity to MF, although there is evidence of the involvement of the OCT2 in MF transfer into the target cells, including the ovarian cells [308].
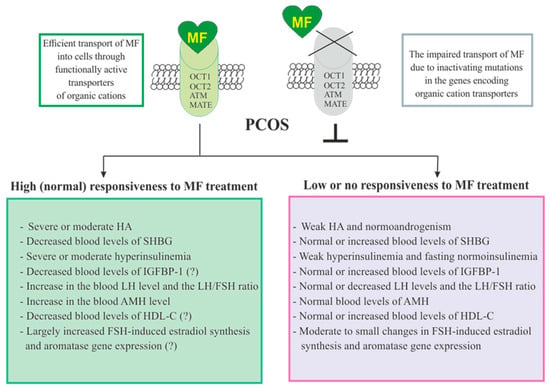
Figure 3. Factors determining responsiveness to metformin and the effectiveness of metformin therapy in women with PCOS. Women with PCOS, as well as the patients with other pathologies, must have functionally active transporters of organic cations (OCT1, OCT2, and others) in order to respond to MF, since inactivating mutations and polymorphisms in the genes encoding these transporters lead to impairment of MF transport into the cell and make MF therapy ineffective. Since MF improves metabolic parameters and insulin sensitivity, its effectiveness in PCOS women with overweight or obesity, as well as with severe dyslipidemia and impaired glucose tolerance, is usually higher. There is evidence that MF therapy is most effective in PCOS women who have pronounced signs of hyperinsulinemia and hyperandrogenism, the increased LH levels and the LH/FSH ratio, the decreased levels of SHBG, IGFBP-1 and HDL-C, and the increased levels of AMH. It can also be assumed that MF will be more effective in patients with increased aromatase expression and ovarian hypersensitivity to FSH, since one of the mechanisms of MF action is normalization of the expression of genes encoding the FSH receptor and aromatase, as well as normalization in the response of ovarian cells to stimulation of FSH. Details and bibliographic references are presented in the Section 2.5. Abbreviations: AMH, anti-Müllerian hormone; FSH, follicle-stimulating hormone; HDL-C, high-density lipoprotein cholesterol; IGFBP-1, insulin-like growth factor-binding protein-1; LH, luteinizing hormone; OCT1 and OCT2, organic cation transporters-1 and 2; SHBG, androgen and sex hormone-binding globulin.
In addition to negative effects for MF transport into the cell, a number of other factors contribute the sensitivity of PCOS patients to MF therapy. Among these factors are the severity of hyperinsulinemia and HA, the blood levels of the binding proteins for insulin, IGF-1 (IGFBP-1) and androgens (SHBG), and the blood levels of AMH and HDL-C (Figure 3).
3. Metformin and Gestational Diabetes Mellitus
Over the past years, the focus has been on the validity of the use of MF for the treatment of GDM and the prevention of GDM-associated preeclampsia, which is of great importance for a normal pregnancy and the birth of healthy offspring [309,310,311,312,313,314,315,316,317,318]. GDM is recognized on average in 10–20% of pregnant women, and the incidence is largely depends on diagnostic criteria and genetic, environmental and ethnic factors [319,320,321]. The dietary and exercise interventions are often used to prevent and treat GDM and improve pregnancy [317,322,323], but such interventions are effective only when used concurrently, and with pharmacological intervention [322,324]. Combining diet and exercise with vitamin D and myo-inositol supplement has been shown to be effective [324,325,326,327]. The most widely used pharmacological approach for GDM correction is insulin therapy, which is currently considered the “gold standard” of care in GDM [317,328,329]. Notably, insulin treatment leads to a number of side effects, primarily hypoglycemic episodes, dangerous for the health of pregnant women and fetus. As a result, the development of alternative pharmacological approaches is now underway, among which the use of MF and other oral hypoglycemic agents is of the greatest interest [317,330,331,332].
In 2007–2013, the first series of randomized clinical trials was carried out to assess the efficacy and safety of MF therapy in GDM, including the comparison with insulin therapy [333,334,335,336,337,338]. It was concluded that MF, to a greater extent than insulin, reduces body weight gain in pregnant women, and also reduces the frequency and severity of gestational hypertension. Unlike insulin, MF did not induce hypoglycemic episodes in the mother and fetus. At the same time, MF therapy had a little or no effect on the incidence of small for gestational age (SGA) or large for gestational-age (LGA) neonates, and had a little effect on the incidence of preeclampsia.
Subsequently, there were the additional evidences of the effectiveness of MF as a drug that reduces maternal body weight and prevents gestational hypertension [314,339,340]. Based on a large clinical data [337,341,342,343,344,345,346,347,348,349], it was concluded that MF in GDM and T2DM, to a greater extent than insulin, prevents neonatal hypoglycemia, and significantly reduces the incidence of fetuses with LGA, thereby reducing the number of newborns who require intensive care after birth [321,339,350]. It is important to note that MF therapy does not increase the rate of preterm delivery, Cesarean section, and SGA neonates [321,339,340], despite the fact that earlier studies showed the risks of MF therapy for a normal pregnancy in GDM [334,351]. Polish scientists found some increase in triglyceride levels and atherogenic index in the third trimester of pregnancy in MF-treated women with GDM. At the same time, they indicated that short-term treatment with MF and insulin had a similar impact on lipid markers of MetS in women with GDM [352].
Several advantages of MF therapy in GDM treatment were demonstrated when comparing MF efficiency with that of other oral hypoglycemic agents, including glibenclamide and its analogues [330,347,353,354,355]. The use of glyburide, which, like MF, provides adequate glycemic control in GDM, is associated with an increased risk of neonatal hypoglycemia, high neonatal birth weight and macrosomia [330,353,355].
As noted above, there is evidence that MF reduces preeclampsia in GDM, and this is based on the ability of MF to prevent endothelial dysfunctions via normalizing the levels of the anti- and pro-angiogenic factors and improving the mitochondrial energy and biogenesis [313,356,357]. MF decreases the production of anti-angiogenic factors, including soluble forms of receptors of angiogenic factors. Among them, there are the placenta-produced soluble vascular endothelial growth factor receptor-1 (VEGFR-1), also called soluble fms-like tyrosine kinase-1 (sFlt-1), which specifically binds to vascular endothelial growth factor-A (VEGF-A) and placental growth factor (PlGF), and the soluble endoglin, a soluble isomer of endoglin, which inhibits the specific binding of TGF-β1 to its receptor [313,356,357,358]. Moreover, MF and its combination with other drugs (esomeprazole, sulfasalazine) lead to an increase in the expression of VEGF-A and placental growth factor (PlGF), both powerful activators of angiogenesis, and to a decrease in TNFα-induced expression of endothelin-1, a potent vasoconstrictor [356,357]. In pregnant women, the increased activity of anti-angiogenic factors induces the systemic endothelial dysfunctions and vasospasm and provokes preeclampsia [358]. MF treatment of mice with a preeclampsia-like model induced by a six-week high-fat diet (HFD) before pregnancy reduces the blood pressure, prevents the proteinuria, normalizes the fetal and maternal weight, and leads to an improvement of the placental labyrinth and fetal vascular development, impaired in the conditions of preeclampsia [359]. This is based on MF-induced normalization of placental production of MMP-2 and VEGF in preeclamptic mice, which, in turn, improves the pregnancy outcomes [359].
In GDM, MF-induced normalization of angiogenesis and blood circulation prevents hypoperfusion of the placenta. In this regard, it should be noted that hypoperfusion leads to placental hypoxia and ischemia and oxidative stress at the maternal-fetal interface, and then the inflammatory factors and the oxidized and degraded biomolecules are transferred into the mother’s bloodstream, causing preeclamptic symptoms [360,361]. Another mechanism for the prevention of preeclampsia may be a MF-induced decrease in IR and normalization of insulin levels in women with GDM, since there is an evidence that a decrease in insulin sensitivity is an important factor provoking preeclampsia [362,363].
It is also assumed that in GDM the protective effect of MF on preeclampsia may be due to its effect on the sensitivity of the placenta to human chorionic gonadotropin (hCG) during the early pregnancy. hCG is the most important regulator of placental development and angiogenesis and is able to influence the ratio of PlGF and sFlt-1 receptors in the blood and, thereby, control the activity of pro- and anti-angiogenic factors [364]. During early pregnancy, high levels of hCG are associated with the elevated sFlt-1/PlGF ratio and the increased risk of preeclampsia [364]. The pregnant women with GDM are more likely to deliver by Cesarean section. The success of Cesarean section is significantly increased with the combined use of MF and insulin and is higher than with the use of insulin or MF alone [365]. The combination MF plus insulin had an improving effect on the delivery of LGA infant as compared to insulin monotherapy. These data indicate the need for the combined use of MF and insulin in treating GDM women with high-risk pregnancy and recommendation for Cesarean section [365].
When using MF to treat GDM, it is important to take into account the ability of this drug to easily cross the placenta barrier to affect the embryo and developing fetus [366]. At the same time, the available data do not support the teratogenic effect of MF when used during pregnancy [315,345,346,351,353,367,368,369,370,371]. Moreover, a number of positive effects of prenatally administered MF on fetal development and functional indices of newborns in GDM were demonstrated [366,372,373]. In GDM, the side effects of MF, as in the treatment of PCOS and T2DM, are mainly limited to gastrointestinal dysfunctions [287,334,374,375].
The fact that, in GDM, MF therapy in about half of cases has little or no effect [334,376], may be due to the genetic and ethnically defined environmental ethnic features, as well as the severity and pattern of hormonal and metabolic disorders in women with GDM. In this regard, the situation is similar to that in PCOS women, where the factors that determine the effectiveness of MF therapy are hyperinsulinemia, HA, functional activity of the gonadotropin and insulin/IGF-1 signaling systems in the ovaries, lipids spectrum, expression and activity of inflammatory factors and antioxidant enzymes, and also organic cation transporter-dependent transfer of MF into the target cells (see Figure 3).
To fully assess the response of pregnant women with GDM to MF, the effect of this drug on glucose homeostasis should be investigated. The glucose level one hour after the start of the oral glucose tolerance test above 212 mg/dL and the mean fasting glucose level during the first week of MF therapy above 95 mg/dL are both reliable indices for supplementing MF therapy with insulin [376]. The differences in responsiveness to MF therapy are believed to explain the results for a number of recent studies and meta-analyzes that did not show a significant effect of MF on pregnancy and its outcomes in women with GDM and on the development of preeclampsia. Based on this, a number of authors have expressed the opinion that insulin is preferable to MF in GDM treatment [377,378,379].
4. Metformin Treatment of Women with Diabetes Mellitus and Obesity
MF, the main oral hypoglycemic agent for the treatment of T2DM, is also widely used to treat women with T2DM before pregnancy. At the onset of pregnancy, for a significant proportion of diabetic women, on average from 11.4% to 62.5%, MF therapy is replaced by insulin therapy, which is still considered more acceptable for glycemic control in pregnancy [380,381]. However, with the accumulation of clinical data, the validity increases for the use of MF to treat women with T2DM during pregnancy, mainly due to the optimization of strategy of MF use and strong evidence of its prenatal safety [341,343,346,349,370,375,381,382,383,384,385,386,387].
As in the case of PCOS and GDM, the effectiveness of MF in the treatment of pregnant women with T2DM depends on a number of factors, including individual susceptibility to this drug. Reduced response to MF requires the use of insulin supplements to improve glycemic control. The number of pregnant T2DM women receiving MF treatment, which require supplementary insulin therapy varied significantly and ranged widely from 4% [385] to 43% [341] and even up to 84% [346]. Supplementary insulin therapy usually begins on average after the 25th week of gestation [346]. The need to add insulin is due to the ineffectiveness of glycemic control using MF alone, even with a significant increase in the daily doses of this drug. Egyptian scientists have shown that in 37% and 39% of pregnant women with T2DM, MF in the daily doses of 1500 and 2000 mg, respectively, provided a good glycemic control, while in 24% of pregnant diabetic women the use of a daily dose of 2000 mg did not secure reliable glycemic control, thus these patients required supplementary insulin treatment [343].
In contrast to women with T2DM, the use of MF in obese pregnant women who do not have severe IR, hyperglycemia and dyslipidemia is not effective. Most clinical studies indicate that MF does not significantly affect the pregnancy and the health of newborns in obese pregnant women [318,388,389,390]. The use of MF in pregnant women with overweight or obesity at the end of the first and beginning of the second trimesters does not significantly improve the outcomes of pregnancy and childbirth [390]. Although some meta-analyzes show that MF treatment of obese women can reduce maternal gestational weight gain and prevent preeclampsia, without negatively affecting the fetus, including stillbirth and congenital abnormalities [314,388], there is currently no reliable reason to recommend MF for the prevention of adverse pregnancy outcomes in obese or overweight women without pronounced signs of T2DM [388,390].
Women with type 1 diabetes mellitus (T1DM) are also characterized by an increased risk of developing preeclampsia and adverse pregnancy outcomes, and these risks are significantly higher than those in T2DM [391,392,393,394,395]. In this regard, it becomes necessary to reduce these risks, for which, traditionally, insulin therapy is used, both in the period before and during pregnancy. Adequate insulin therapy for women with T1DM before pregnancy reduces the risk of hypertensive disorder of pregnancy [396].
However, a long-term therapy with increasing doses of insulin leads to IR and hypoglycemic episodes, and also increases the mother’s body weight, thereby worsening the outcome of pregnancy [397,398,399]. In the third trimester of pregnancy, the daily insulin dose can increase to 1.0 IU/kg, and the increase in this dose can occur rapidly [400]. Weight gain in pregnant women with T1DM, which is more pronounced than among non-diabetic patients, and persistent hyperglycemia lead to a higher risk of fetal overgrowth, resulting in LGA and macrosomia [401,402].
Optimization of insulin therapy using automated insulin delivery is one of the approaches to prevent IR and normalize the body weight and glucose homeostasis [399,403,404]. Another, alternative, approach is the use of MF therapy to correct hyperglycemia in pregnant women with T1DM, especially since MF has been successfully used for a long period of time to correct hyperglycemia in non-pregnant patients with T1DM, which makes it possible to increase insulin sensitivity and reduce the insulin dose [405,406,407,408,409]. The data on MF treatment of pregnant women with T1DM are encouraging, demonstrating improved glycemic control without the need to increase the insulin dose and reducing maternal weight gain and the risk of neonatal hypoglycemia [410].
5. Conclusions
The data presented in the review convincingly prove that MF has an improving effect on reproductive functions in women with PCOS, GDM and T2DM. At the same time, the effectiveness of MF therapy is due to a large number of different factors that must be taken into account when choosing this therapy and also when developing a strategy for using MF. Firstly, it is necessary to assess the efficiency of MF transport into the cell, which depends on the functional activity of the organic cation transporters and can be disrupted by inactivating mutations in their genes. The presence of certain mutations leads to a loss of responsiveness to MF and makes the use of MF therapy meaningless. Secondly, MF therapy is more effective in the severely overweight and obese patients with IR, compensatory hyperinsulinemia, impaired glucose tolerance, as well as with dyslipidemia, which is due to a decrease in the blood levels of HDL-C. This is not surprising, since the clinical effect of MF therapy is due to an improvement in insulin sensitivity, a decrease in the adipose tissue mass, and restoration of the glucose and lipid metabolism.
In addition, as demonstrated in a number of clinical studies in PCOS women, the effectiveness of MF is largely determined by the hormonal status of the ovaries and the functioning of the HPG axis. The MF therapy may be most effective in PCOS women who have: (1) severe HA, which may be due to hyperactivation of ovarian insulin/IGF-1-regulated signaling pathways that stimulate androgen synthesis, as well as an increase in LH levels and the LH/FSH ratio and a decrease in the blood levels of IGFBP-1 and SHBG; (2) an increase in the AMH production; and (3) an increased aromatase expression and FSH-induced estrogen synthesis in the ovaries. There is reason to believe that various combinations of these factors, including those with IR and metabolic disorders, may become reliable indications for prescribing MF alone and in combinations with other drugs, diet or exercises to correct the reproductive functions in PCOS. This can be helpful when using MF to treat the pregnant women with GDM and T2DM.
Since some of MF targets overlap well with those of leptin, the assessment of leptin status in patients with reproductive disorders may also be important. As a result, leptin resistance, both systemic and in the ovaries, as well as the changes in the hypothalamic leptin signaling pathways, negatively affecting the production of GnRH, can become factors that will determine the effectiveness of MF therapy. In this regard, it should be noted that the central mechanisms of action of MF, which easily penetrates the CNS and improves the metabolism of the neuronal and glial cells, still remain underestimated. By acting on the CNS, MF restores the signaling networks of the hypothalamus and the other brain regions that are involved in the control of reproductive functions and undergo significant compensatory and pathological changes in metabolic and endocrine disorders, including PCOS, GDM, T2DM, and MetS.
A unique feature of MF is the multiplicity of molecular mechanisms of its action on target cells, which include direct or indirect regulation of the AMPK-, calcium- and cAMP-dependent signaling pathways, as well as the MAPK cascade and the IRS/PI 3-K/Akt pathway. As a result, MF controls not only energy and metabolic processes in the cell, but also the processes of growth, differentiation, apoptosis, inflammation, and ER stress. At the same time, most of the regulatory effects of MF are based largely on its modulating and normalizing influence on intracellular signaling cascades than on their prolonged stimulation or suppression. Depending on the functional state of the IRS/PI 3-K/Akt pathway, MF can either prevent its hyperactivation, which is especially important for its antitumor effect, or, on the contrary, restore its reduced activity, improving the survival of target cells and their sensitivity to insulin and leptin. As expected, MF therapy affects the responsiveness of hypothalamic neurons, pituitary gonadotrophs, and ovarian cells to the hormones, growth factors, adipokines and cytokines, but more studies are required for complete elucidation of all regulatory mechanisms involved.
The use of MF in combination with the other drugs has great potential. This is supported by the encouraging results of clinical trials of the combined therapy with MF and insulin in pregnant women with GDM and T2DM. In metabolic and endocrine disorders, the combined therapy not only allows to increase the efficiency and pattern of the effects of MF on the HPG axis, but also to reduce the pharmacological doses of drugs, including MF, thus avoiding possible side effects of high-dose drug administration, including the undesirable effect of MF on the functioning of the gastrointestinal tract.
The presented results indicate a significant and not yet fully understood potential of MF therapy for the correction of reproductive dysfunctions in women. Significantly, of great importance are the absence of a teratogenic effect of MF and the low risks of MF therapy on the health of the mother and child. It should be taken into account that the unjustified use of MF for the treatment of patients lacking profoundly manifesting metabolic and endocrine disorders can lead to energy and metabolic imbalance and further deterioration of the functional state of their reproductive system.
This entry is adapted from the peer-reviewed paper 10.3390/ph14010042
References
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585, doi:10.1007/s00125-017-4342-z.
- Li, M.; Li, X.; Zhang, H.; Lu, Y. Molecular Mechanisms of Metformin for Diabetes and Cancer Treatment. Front. Physiol. 2018, 9, 1039, doi:10.3389/fphys.2018.01039.
- Agius, L.; Ford, B.E.; Chachra, S.S. The Metformin Mechanism on Gluconeogenesis and AMPK Activation: The Metabolite Perspective. Int. J. Mol. Sci. 2020, 21, 3240, doi:10.3390/ijms21093240.
- An, H.; He, L. Current understanding of metformin effect on the control of hyperglycemia in diabetes. J. Endocrinol. 2016, 228, R97–R106, doi:10.1530/JOE-15-0447.
- He, L. Metformin and Systemic Metabolism. Trends Pharm. Sci. 2020, 41, 868–881, doi:10.1016/j.tips.2020.09.001.
- Cioce, M.; Pulito, C.; Strano, S.; Blandino, G.; Fazio, V.M. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells 2020, 9, 2439, doi:10.3390/cells9112439.
- Chan, P.; Shao, L.; Tomlinson, B.; Zhang, Y.; Liu, Z.M. Metformin transporter pharmacogenomics: Insights into drug dispo-sition-where are we now? Expert Opin. Drug Metab. Toxicol. 2018, 14, 1149–1159, doi:10.1080/17425255.2018.1541981
- Lee, N.; Hebert, M.F.; Wagner, D.J.; Easterling, T.R.; Liang, C.J.; Rice, K.; Wang, J. Organic Cation Transporter 3 Facilitates Fetal Exposure to Metformin during Pregnancy. Mol. Pharmacol. 2018, 94, 1125–1131, doi:10.1124/mol.118.112482.
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262, doi:10.1038/nrm3311.
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 27, 190–201, doi:10.1016/j.tcb.2015.10.013.
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313, doi:10.1016/j.cmet.2017.10.009.
- Hardie, D.G. AMPK: A key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. 2008, 32, S7–S12, doi:10.1038/ijo.2008.116.
- Hardie, DG. Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface. 2018, 15, 20170774, doi:10.1098/rsif.2017.0774.
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843, doi:10.1038/sj.emboj.7600110
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodu-lin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19, doi:10.1016/j.cmet.2005.05.009.
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33, doi:10.1016/j.cmet.2005.06.005.
- Momcilovic, M.; Hong, S.P.; Carlson, M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J. Biol. Chem. 2006, 281, 25336–25343, doi:10.1074/jbc.M604399200.
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199, doi:10.1042/BJ20150910.
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Ga-lectins Control mTOR in Response to Endomembrane Damage. Mol. Cell 2018, 70, 120–135, doi:10.1016/j.molcel.2018.03.009.
- Jia, J.; Bissa, B.; Brecht, L.; Allers, L.; Choi, S.W.; Gu, Y.; Zbinden, M.; Burge, M.R.; Timmins, G.; Hallows, K.; et al. AMPK, a Regulator of Metabolism and Autophagy, Is Activated by Lysosomal Damage via a Novel Galectin-Directed Ubiquitin Sig-nal Transduction System. Mol. Cell. 2020, 77, 951–969, doi:10.1016/j.molcel.2019.12.028.
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174, doi:10.1172/JCI13505.
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008, doi:10.1016/j.cub.2003.10.031.
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446, doi:10.1074/jbc.M114.567271.
- Oakhill, J.S.; Steel, R.; Chen, Z.P.; Scott, J.W.; Ling, N.; Tam, S.; Kemp, B.E. AMPK is a direct adenylate charge-regulated pro-tein kinase. Science 2011, 332, 1433–1435, doi:10.1126/science.1200094.
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233, doi:10.1038/nature09932.
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fruc-tose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116, doi:10.1038/nature23275.
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical activa-tion of compartmentalized pools of AMPK depends on severity of nutrient or energy stress. Cell Res. 2019, 29, 460–473, doi:10.1038/s41422-019-0163-6.
- Davies, S.P.; Helps, N.R.; Cohen, P.T.; Hardie, D.G. 5’-AMP inhibits dephosphorylation, as well as promoting phosphoryla-tion, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and na-tive bovine protein phosphatase-2AC. FEBS Lett. 1995, 377, 421–425, doi:10.1016/0014-5793(95)01368-7.
- Suter, M.; Riek, U.; Tuerk, R.; Schlattner, U.; Wallimann, T.; Neumann, D. Dissecting the role of 5’-AMP for allosteric stimu-lation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006, 281, 32207–32216, doi:10.1074/jbc.M606357200.
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228, doi:10.1074/jbc.275.1.223.
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of com-plex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614, doi:10.1042/bj3480607.
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metfor-min inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369, doi:10.1172/JCI40671.
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487, doi:10.1042/BJ20140620.
- Ouyang, J.; Parakhia, R.A.; Ochs, R.S. Metformin activates AMP kinase through inhibition of AMP deaminase. J. Biol. Chem. 2011, 286, 1–11, doi:10.1074/jbc.M110.121806.
- Meng, S.; Cao, J.; He, Q.; Xiong, L.; Chang, E.; Radovick, S.; Wondisford, F.E.; He, L. Metformin activates AMP-activated protein kinase by promoting formation of the αβγ heterotrimeric complex. J. Biol. Chem. 2015, 290, 3793–3802, doi:10.1074/jbc.M114.604421.
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569.
- Sliwinska, A.; Drzewoski, J. Molecular action of metformin in hepatocytes: An updated insight. Curr. Diabetes Rev. 2015, 11, 175–181, doi:10.2174/1573399811666150325233108.
- Karnewar, S.; Neeli, P.K.; Panuganti, D.; Kotagiri, S.; Mallappa, S.; Jain, N.; Jerald, M.K.; Kotamraju, S. Metformin regulates mitochondrial biogenesis and senescence through AMPK mediated H3K79 methylation: Relevance in age-associated vascular dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1115–1128, doi:10.1016/j.bbadis.2018.01.018.
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispen-sable manner. J. Cell. Mol. Med. 2011, 15, 166–178, doi:10.1111/j.1582-4934.2009.00954.x
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing ef-fects of metformin. Nat. Med. 2013, 19, 1649–1654, doi:10.1038/nm.3372.
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224, doi:10.1038/emm.2016.16.
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J. Cell. Mol. Med. 2011, 15, 166–178, doi:10.1111/j.1582-4934.2009.00954.x
- Motoshima, H.; Goldstein, B.J.; Igata, M.; Araki, E. AMPK and cell proliferation—AMPK as a therapeutic target for atherosclerosis and cancer. J. Physiol. 2006, 574, 63–71, doi:10.1113/jphysiol.2006.108324.
- Choi, Y.K.; Park, K.G. Metabolic roles of AMPK and metformin in cancer cells. Mol. Cells 2013, 36, 279–87, doi:10.1007/s10059-013-0169-8.
- Gao, F.; Chen, J.; Zhu, H. A potential strategy for treating atherosclerosis: Improving endothelial function via AMP-activated protein kinase. Sci. China Life Sci. 2018, 61, 1024–1029, doi:10.1007/s11427-017-9285-1.
- Lyons, C.L.; Roche. H.M. Nutritional Modulation of AMPK-Impact upon Metabolic-Inflammation. Int. J. Mol. Sci. 2018, 19, 3092, doi:10.3390/ijms19103092.
- Viollet, B.; Foretz, M. Revisiting the mechanisms of metformin action in the liver. Ann. Endocrinol. 2013, 74, 123–129, doi:10.1016/j.ando.2013.03.006.
- Johanns, M.; Lai, Y.C.; Hsu, M.F.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.E.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856, doi:10.1038/ncomms10856.
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by de-creasing production of cyclic AMP. Nature 2013, 494, 256–260, doi:10.1038/nature11808.
- He, L.; Sabet, A.; Djedjos, S.; Miller, R.; Sun, X.; Hussain, M.A.; Radovick, S.; Wondisford, F.E. Metformin and insulin sup-press hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell 2009, 137, 635–646, doi:10.1016/j.cell.2009.03.016.
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogen-ase. Nature 2014, 510, 542–546, doi:10.1038/nature13270.
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.M.; Zhang, D.; Camporez, J.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med. 2018, 24, 1384–1394, doi:10.1038/s41591-018-0125-4.
- Cuyàs, E.; Verdura, S.; Llorach-Pares, L.; Fernández-Arroyo, S.; Luciano-Mateo, F.; Cabré, N.; Stursa, J.; Werner, L.; Mar-tin-Castillo, B.; Viollet, B.; et al. Metformin directly targets the H3K27me3 demethylase KDM6A/UTX. Aging Cell. 2018, 17, e12772, doi:10.1111/acel.12772.
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contrib-uting to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858, doi:10.1038/nm.4345.
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735, doi:10.1136/gutjnl-2012-303839.
- Duca, F.A.; Côté, C.D.; Rasmussen, B.A.; Zadeh-Tahmasebi, M.; Rutter, G.A.; Filippi, B.M.; Lam, T.K. Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med. 2015, 21, 506–511, doi:10.1038/nm.3787.
- Hattori, Y.; Suzuki, K.; Hattori, S.; Kasai, K. Metformin inhibits cytokine-induced nuclear factor kappaB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 2006, 47, 1183–1188, doi:10.1161/01.HYP.0000221429.94591.72.
- Huang, N.L.; Chiang, S.H.; Hsueh, C.H.; Liang, Y.J.; Chen, Y.J.; Lai, L.P. Metformin inhibits TNF-alpha-induced IkappaB ki-nase phosphorylation, IkappaB-alpha degradation and IL-6 production in endothelial cells through PI3K-dependent AMPK phosphorylation. Int. J. Cardiol. 2009, 134, 169–175, doi:10.1016/j.ijcard.2008.04.010.
- Okamura, H.; Yoshida, K.; Sasaki, E.; Qiu, L.; Amorim, B.R.; Morimoto, H.; Haneji, T. Expression of PTEN and Akt phos-phorylation in lipopolysaccharide-treated NIH3T3 cells. Cell Biol. Int. 2007, 31, 119–125, doi:10.1016/j.cellbi.2006.09.014.
- Lee, S.K.; Lee, J.O.; Kim, J.H.; Kim, S.J.; You, G.Y.; Moon, J.W.; Jung, J.H.; Park, S.H.; Uhm, K.O.; Park, J.M.; et al. Metformin sensitizes insulin signaling through AMPK-mediated PTEN down-regulation in preadipocyte 3T3-L1 cells. J. Cell. Biochem. 2011, 112, 1259–1267, doi:10.1002/jcb.23000.
- Balen, A.H.; Morley, L.C.; Misso, M.; Franks, S.; Legro, R.S.; Wijeyaratne, C.N.; Stener-Victorin, E.; Fauser, B.C.; Norman, R.J.; Teede, H. The management of anovulatory infertility in women with polycystic ovary syndrome: An analysis of the evidence to support the development of global WHO guidance. Hum. Reprod. Update 2016, 22, 687–708, doi:10.1093/humupd/dmw025.
- Norman, R.J.; Dewailly, D.; Legro, R.S.; Hickey, T.E. Polycystic ovary syndrome. Lancet 2007, 370, 685–697, doi:10.1016/S0140-6736(07)61345-2.
- Fauser, B.C.; Tarlatzis, B.C.; Rebar, R.W.; Legro, R.S.; Balen, A.H.; Lobo, R.; Carmina, E.; Chang, J.; Yildiz, B.O.; Laven, J.S.; et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): The Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil. Steril. 2012, 97, 28–38.e25, doi:10.1016/j.fertnstert.2011.09.024.
- Abbott, D.H.; Dumesic, D.A.; Levine, J.E. Hyperandrogenic origins of polycystic ovary syndrome—Implications for patho-physiology and therapy. Expert Rev. Endocrinol. Metab. 2019, 14, 131–143, doi:10.1080/17446651.2019.1576522.
- Witchel, S.F.; Oberfield, S.E.; Peña, A.S. Polycystic Ovary Syndrome: Pathophysiology, Presentation, and Treatment with Emphasis on Adolescent Girls. J. Endocr. Soc. 2019, 3, 1545–1573, doi:10.1210/js.2019-00078.
- Alvarez-Blasco, F.; Botella-Carretero, J.I.; San Millán, J.L.; Escobar-Morreale, H.F. Prevalence and characteristics of the pol-ycystic ovary syndrome in overweight and obese women. Arch. Intern. Med. 2006, 166, 2081–2086, doi:10.1001/archinte.166.19.2081.
- Legro, R.S.; Barnhart, H.X.; Schlaff, W.D.; Carr, B.R.; Diamond, M.P.; Carson, S.A.; Steinkampf, M.P.; Coutifaris, C.; McGov-ern, P.G.; Cataldo, N.A.; et al. Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N. Engl. J. Med. 2007, 356, 551–566, doi:10.1056/NEJMoa063971.
- Azziz, R.; Carmina, E.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.F.; Futterweit, W.; Janssen, O.E.; Legro, R.S.; Norman, R.J.; Taylor, A.E.; et al. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: The complete task force report. Fertil. Steril. 2009, 91, 456–488, doi:10.1016/j.fertnstert.2008.06.035.
- Li, R.; Zhang, Q.; Yang, D.; Li, S.; Lu, S.; Wu, X.; Wei, Z.; Song, X.; Wang, X.; Fu, S.; et al. Prevalence of polycystic ovary syn-drome in women in China: A large community-based study. Hum. Reprod. 2013, 28, 2562–2569, doi:10.1093/humrep/det262.
- Kollmann, M.; Klaritsch, P.; Martins, W.P.; Guenther, F.; Schneider, V.; Herzog, S.A.; Craciunas, L.; Lang, U.; Obermay-er-Pietsch, B.; Lerchbaum, E.; et al. Maternal and neonatal outcomes in pregnant women with PCOS: Comparison of differ-ent diagnostic definitions. Hum. Reprod. 2015, 30, 2396–2403, doi:10.1093/humrep/dev187.
- Cassar, S.; Misso, M.L.; Hopkins, W.G.; Shaw, C.S.; Teede, H.J.; Stepto, N.K. Insulin resistance in polycystic ovary syndrome: A systematic review and meta-analysis of euglycaemic-hyperinsulinaemic clamp studies. Hum. Reprod. 2016, 31, 2619–2631, doi:10.1093/humrep/dew243.
- Spritzer, P.M. Polycystic ovary syndrome: Reviewing diagnosis and management of metabolic disturbances. Arq. Bras. En-docrinol. Metabol. 2014, 58, 182–187, doi:10.1590/0004-2730000003051.
- Dumesic, D.A.; Oberfield, S.E.; Stener-Victorin, E.; Marshall, J.C.; Laven, J.S.; Legro, R.S. Scientific Statement on the Diagnos-tic Criteria, Epidemiology, Pathophysiology, and Molecular Genetics of Polycystic Ovary Syndrome. Endocr. Rev. 2015, 36, 487–525, doi:10.1210/er.2015-1018.
- Faure, M.; Bertoldo, M.J.; Khoueiry, R.; Bongrani, A.; Brion, F.; Giulivi, C.; Dupont, J.; Froment, P. Metformin in Reproduc-tive Biology. Front. Endocrinol. 2018, 9, 675, doi:10.3389/fendo.2018.00675.
- Jones, M.R.; Goodarzi, M.O. Genetic determinants of polycystic ovary syndrome: Progress and future directions. Fertil. Steril. 2016, 106, 25–32, doi:10.1016/j.fertnstert.2016.04.040.
- Liu, H.; Zhao, H.; Chen, Z.J. Genome-Wide Association Studies for Polycystic Ovary Syndrome. Semin. Reprod. Med. 2016, 34, 224–229, doi:10.1055/s-0036-1585403.
- Khan, M.J.; Ullah, A.; Basit, S. Genetic Basis of Polycystic Ovary Syndrome (PCOS): Current Perspectives. Appl. Clin. Genet. 2019, 12, 249–260, doi:10.2147/TACG.S200341.
- Dapas, M.; Lin, F.T.J.; Nadkarni, G.N.; Sisk, R.; Legro, R.S.; Urbanek, M.; Hayes, M.G.; Dunaif, A. Distinct subtypes of poly-cystic ovary syndrome with novel genetic associations: An unsupervised, phenotypic clustering analysis. PLoS Med. 2020, 17, e1003132, doi:10.1371/journal.pmed.1003132.
- Xita, N.; Tsatsoulis, A. Review: Fetal programming of polycystic ovary syndrome by androgen excess: Evidence from ex-perimental, clinical, and genetic association studies. J. Clin. Endocrinol. Metab. 2006, 91, 1660–1666, doi:10.1210/jc.2005-2757.
- Ilie, I.R.; Georgescu, C.E. Polycystic Ovary Syndrome-Epigenetic Mechanisms and Aberrant MicroRNA. Adv. Clin. Chem. 2015, 71, 25–45, doi:10.1016/bs.acc.2015.06.001.
- Yu, Y.Y.; Sun, C.X.; Liu, Y.K.; Li, Y.; Wang, L.; Zhang, W. Genome-wide screen of ovary-specific DNA methylation in poly-cystic ovary syndrome. Fertil. Steril. 2015, 104, 145–153, doi:10.1016/j.fertnstert.2015.04.005.
- Wang, S.; Alvero, R. Racial and ethnic differences in physiology and clinical symptoms of polycystic ovary syndrome. Semin. Reprod. Med. 2013, 31, 365–369, doi:10.1055/s-0033-1348895.
- Merkin, S.S.; Phy, J.L.; Sites, C.K.; Yang, D. Environmental determinants of polycystic ovary syndrome. Fertil. Steril. 2016, 106, 16–24, doi:10.1016/j.fertnstert.2016.05.011.
- Abdolahian, S.; Tehrani, F.R.; Amiri, M.; Ghodsi, D.; Yarandi, R.B.; Jafari, M.; Majd, H.A.; Nahidi, F. Effect of lifestyle modi-fications on anthropometric, clinical, and biochemical parameters in adolescent girls with polycystic ovary syndrome: A systematic review and meta-analysis. BMC Endocr. Disord. 2020, 20, 71, doi:10.1186/s12902-020-00552-1.
- Abbott, D.H.; Barnett, D.K.; Bruns, C.M.; Dumesic, D.A. Androgen excess fetal programming of female reproduction: A de-velopmental etiology for polycystic ovary syndrome? Hum. Reprod. Update 2005, 11, 357–374, doi:10.1093/humupd/dmi013.
- Abbott, D.H.; Bacha, F. Ontogeny of polycystic ovary syndrome and insulin resistance in utero and early childhood. Fertil. Steril. 2013, 100, 2–11, doi:10.1016/j.fertnstert.2013.05.023.
- de Melo, A.S.; Dias, S.V.; Cavalli, Rde.C.; Cardoso, V.C.; Bettiol, H.; Barbieri, M.A.; Ferriani, R.A.; Vieira, C.S. Pathogenesis of polycystic ovary syndrome: Multifactorial assessment from the foetal stage to menopause. Reproduction 2015, 150, 1–24, doi:10.1530/REP-14-0499.
- Morley, L.C.; Tang, T.; Yasmin, E.; Norman, R.J.; Balen, A.H. Insulin-sensitising drugs (metformin, rosiglitazone, pioglita-zone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst. Rev. 2017, 11, CD003053, doi:10.1002/14651858.CD003053.pub6.
- Practice Committee of the American Society for Reproductive Medicine. Electronic address: ASRM@asrm.org; Practice Committee of the American Society for Reproductive Medicine. Role of metformin for ovulation induction in infertile pa-tients with polycystic ovary syndrome (PCOS): A guideline. Fertil. Steril. 2017, 108, 426–441, doi:10.1016/j.fertnstert.2017.06.026.
- Abdalla, M.A.; Deshmukh, H.; Atkin, S.; Sathyapalan, T. A review of therapeutic options for managing the metabolic aspects of polycystic ovary syndrome. Ther. Adv. Endocrinol. Metab. 2020, 11, 2042018820938305, doi:10.1177/2042018820938305.
- Bordewijk, E.M.; Nahuis, M.; Costello, M.F.; Van der Veen, F.; Tso, L.O.; Mol, B.W.; van Wely, M. Metformin during ovula-tion induction with gonadotrophins followed by timed intercourse or intrauterine insemination for subfertility associated with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2017, 1, CD009090, doi:10.1002/14651858.CD009090.pub2.
- Sharpe, A.; Morley, L.C.; Tang, T.; Norman, R.J.; Balen, A.H. Metformin for ovulation induction (excluding gonadotrophins) in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2019, 12, CD013505, doi:10.1002/14651858.CD013505.
- Gadalla, M.A.; Norman, R.J.; Tay, C.T.; Hiam, D.S.; Melder, A.; Pundir, J.; Thangaratinam, S.; Teede, H.J.; Mol, B.W.J.; Moran, L.J. Medical and Surgical Treatment of Reproductive Outcomes in Polycystic Ovary Syndrome: An Overview of Systematic Reviews. Int. J. Fertil. Steril. 2020, 13, 257–270, doi:10.22074/ijfs.2020.5608.
- Wu, Y.; Tu, M.; Huang, Y.; Liu, Y.; Zhang, D. Association of Metformin With Pregnancy Outcomes in Women With Polycys-tic Ovarian Syndrome Undergoing In Vitro Fertilization: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2011995, doi:10.1001/jamanetworkopen.2020.11995.
- Rojas, J.; Chávez-Castillo, M.; Bermúdez, V. The Role of Metformin in Metabolic Disturbances during Pregnancy: Polycystic Ovary Syndrome and Gestational Diabetes Mellitus. Int. J. Reprod. Med. 2014, 2014, 797681, doi:10.1155/2014/797681.
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; International PCOS Network. Recommendations from the international evidence-based guideline for the assessment and management of pol-ycystic ovary syndrome. Clin. Endocrinol. 2018, 89, 251–268, doi:10.1111/cen.13795.
- Gormsen, L.C.; Søndergaard, E.; Christensen, N.L.; Brøsen, K.; Jessen, N.; Nielsen, S. Metformin increases endogenous glu-cose production in non-diabetic individuals and individuals with recent-onset type 2 diabetes. Diabetologia 2019, 62, 1251–1256, doi:10.1007/s00125-019-4872-7.
- McCreight, L.J.; Mari, A.; Coppin, L.; Jackson, N.; Umpleby, A.M.; Pearson, E.R. Metformin increases fasting glucose clear-ance and endogenous glucose production in non-diabetic individuals. Diabetologia 2020, 63, 444–447, doi:10.1007/s00125-019-05042-1.
- Bryrup, T.; Thomsen, C.W.; Kern, T.; Allin, K.H.; Brandslund, I.; Jørgensen, N.R.; Vestergaard, H.; Hansen, T.; Hansen, T.H.; Pedersen, O.; et al. Metformin-induced changes of the gut microbiota in healthy young men: Results of a non-blinded, one-armed intervention study. Diabetologia 2019, 62, 1024–1035, doi:10.1007/s00125-019-4848-7.
- Derkach, K.V.; Kuznetsova, L.A.; Sharova, T.S.; Ignat’eva, P.A.; Bondareva, V.M.; Shpakov, A.O. The effect of prolonged metformin treatment on the activity of the adenylate cyclase system and NO-synthase in the brain and myocardium of obese rats. Cell Tissue Biol. 2015, 9, 385–394, doi:10.1134/S1990519X1505003X.
- Practice Committee of the American Society for Reproductive Medicine. Electronic address: ASRM@asrm.org; Practice Committee of the American Society for Reproductive Medicine. Role of metformin for ovulation induction in infertile patients with polycystic ovary syndrome (PCOS): A guideline. Fertil. Steril. 2017, 108, 426–441, doi:10.1016/j.fertnstert.2017.06.026.
- Lautatzis, M.E.; Goulis, D.G.; Vrontakis, M. Efficacy and safety of metformin during pregnancy in women with gestational diabetes mellitus or polycystic ovary syndrome: A systematic review. Metabolism 2013, 62, 1522–1534, doi:10.1016/j.metabol.2013.06.006.
- Sivalingam, V.N.; Myers, J.; Nicholas, S.; Balen, A.H.; Crosbie, E.J. Metformin in reproductive health, pregnancy and gynaecological cancer: Established and emerging indications. Hum. Reprod. Update 2014, 20, 853–868, doi:10.1093/humupd/dmu037.
- Feng, L.; Lin, X.F.; Wan, Z.H.; Hu, D.; Du, Y.K. Efficacy of metformin on pregnancy complications in women with polycystic ovary syndrome: A meta-analysis. Gynecol. Endocrinol. 2015, 31, 833–839, doi:10.3109/09513590.2015.1041906.
- Sinai Talaulikar, V.; Tang, T.; Yasmin, E. Role of Metformin in Women’s Health: Review of Its Current Place in Clinical Practice and Emerging Indications for Future. Obstet. Gynecol. Surv. 2016, 71, 307–317, doi:10.1097/OGX.0000000000000312.
- Tan, X.; Li, S.; Chang, Y.; Fang, C.; Liu, H.; Zhang, X.; Wang, Y. Effect of metformin treatment during pregnancy on women with PCOS: A systematic review and meta-analysis. Clin. Invest. Med. 2016, 39, 120–131, doi:10.25011/cim.v39i4.27091.
- Zeng, X.L.; Zhang, Y.F.; Tian, Q.; Xue, Y.; An, R.F. Effects of metformin on pregnancy outcomes in women with polycystic ovary syndrome: A meta-analysis. Medicine 2016, 95, e4526, doi:10.1097/MD.0000000000004526.
- Løvvik, T.S.; Carlsen, S.M.; Salvesen, Ø.; Steffensen, B.; Bixo, M.; Gómez-Real, F.; Lønnebotn, M.; Hestvold, K.V.; Zabielska, R.; Hirschberg, A.L.; Trouva, A.; et al. Use of metformin to treat pregnant women with polycystic ovary syndrome (PregMet2): A randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2019, 7, 256–266, doi:10.1016/S2213-8587(19)30002-6.
- Morley, L.C.; Tang, T.; Yasmin, E.; Norman, R.J.; Balen, A.H. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst. Rev. 2017, 11, CD003053, doi:10.1002/14651858.CD003053.pub6.
- Wu, Y.; Tu, M.; Huang, Y.; Liu, Y.; Zhang, D. Association of Metformin With Pregnancy Outcomes in Women With Polycystic Ovarian Syndrome Undergoing In Vitro Fertilization: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2011995, doi:10.1001/jamanetworkopen.2020.11995.
- Baillargeon, J.P.; Jakubowicz, D.J.; Iuorno, M.J.; Jakubowicz, S.; Nestler, J.E. Effects of metformin and rosiglitazone, alone and in combination, in nonobese women with polycystic ovary syndrome and normal indices of insulin sensitivity. Fertil. Steril. 2004, 82, 893–902, doi:10.1016/j.fertnstert.2004.02.127.
- Carmina, E.; Lobo, R.A. Does metformin induce ovulation in normoandrogenic anovulatory women? Am. J. Obstet. Gynecol. 2004, 191, 1580–1584, doi:10.1016/j.ajog.2004.05.030.
- Bordewijk, E.M.; Nahuis, M.; Costello, M.F.; Van der Veen, F.; Tso, L.O.; Mol, B.W.; van Wely, M. Metformin during ovulation induction with gonadotrophins followed by timed intercourse or intrauterine insemination for subfertility associated with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2017, 1, CD009090, doi:10.1002/14651858.CD009090.pub2.
- Tang, T.; Lord, J.M.; Norman, R.J.; Yasmin, E.; Balen, A.H. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst. Rev. 2010, 1, CD003053, doi:10.1002/14651858.CD003053.pub4.
- Kjøtrød, S.B.; Carlsen, S.M.; Rasmussen, P.E.; Holst-Larsen, T.; Mellembakken, J.; Thurin-Kjellberg, A.; Haapaniemikouru, K.; Morin-Papunen, L.; Humaidan, P.; Sunde, A.; et al. Use of metformin before and during assisted reproductive technology in non-obese young infertile women with polycystic ovary syndrome: A prospective, randomized, double-blind, multi-centre study. Hum. Reprod. 2011, 26, 2045–2053, doi:10.1093/humrep/der154.
- Morin-Papunen, L.; Rantala, A.S.; Unkila-Kallio, L.; Tiitinen, A.; Hippeläinen, M.; Perheentupa, A.; Tinkanen, H.; Bloigu, R.; Puukka, K.; Ruokonen, A.; et al. Metformin improves pregnancy and live-birth rates in women with polycystic ovary syndrome (PCOS): A multicenter, double-blind, placebo-controlled randomized trial. J. Clin. Endocrinol. Metab. 2012, 97, 1492–1500, doi:10.1210/jc.2011-3061.
- Tang, T.; Lord, J.M.; Norman, R.J.; Yasmin, E.; Balen, A.H. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst. Rev. 2012, 5, CD003053, doi:10.1002/14651858.CD003053.pub5.
- Tso, L.O.; Costello, M.F.; Albuquerque, L.E.; Andriolo, R.B.; Macedo, C.R. Metformin treatment before and during IVF or ICSI in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2014, 2014, CD006105, doi:10.1002/14651858.CD006105.pub3.
- Kollmann, M.; Martins, W.P.; Lima, M.L.; Craciunas, L.; Nastri, C.O.; Richardson, A.; Raine-Fenning, N. Strategies for improving outcome of assisted reproduction in women with polycystic ovary syndrome: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2016, 48, 709–718, doi:10.1002/uog.15898.
- Palomba, S.; Falbo, A.; Carrillo, L.; Villani, M.T.; Orio, F.; Russo, T.; Di Cello, A.; Cappiello, F., Capasso, S.; Tolino, A.; et al. Metformin reduces risk of ovarian hyperstimulation syndrome in patients with polycystic ovary syndrome during gonad-otropin-stimulated in vitro fertilization cycles: A randomized, controlled trial. Fertil. Steril. 2011, 96, 1384–1390, doi:10.1016/j.fertnstert.2011.09.020.
- Palomba, S.; Falbo, A.; La Sala, G.B. Effects of metformin in women with polycystic ovary syndrome treated with gonado-trophins for in vitro fertilisation and intracytoplasmic sperm injection cycles: A systematic review and meta-analysis of randomised controlled trials. BJOG 2013, 120, 267–276, doi:10.1111/1471-0528.12070.
- Palomba, S.; Falbo, A.; La Sala, G.B. Effects of metformin in women with polycystic ovary syndrome treated with gonadotrophins for in vitro fertilisation and intracytoplasmic sperm injection cycles: A systematic review and meta-analysis of randomised controlled trials. BJOG 2013, 120, 267–276, doi:10.1111/1471-0528.12070.
- Costello, M.F.; Chapman, M.; Conway, U. A systematic review and meta-analysis of randomized controlled trials on metformin co-administration during gonadotrophin ovulation induction or IVF in women with polycystic ovary syndrome. Hum. Reprod. 2006, 21, 1387–1399, doi:10.1093/humrep/dei501.
- Tso, L.O.; Costello, M.F.; Albuquerque, L.E.; Andriolo, R.B.; Freitas, V. Metformin treatment before and during IVF or ICSI in women with polycystic ovary syndrome. Cochrane Database Syst. Rev. 2009, 2, CD006105, doi:10.1002/14651858.CD006105.pub2.
- Palomba, S.; Falbo, A.; Zullo, F.; Orio, F., Jr. Evidence-based and potential benefits of metformin in the polycystic ovary syndrome: A comprehensive review. Endocr. Rev. 2009, 30, 1–50, doi:10.1210/er.2008-0030.
- Abdalmageed, O.S.; Farghaly, T.A.; Abdelaleem, A.A.; Abdelmagied, A.E.; Ali, M.K.; Abbas, A.M. Impact of Metformin on IVF Outcomes in Overweight and Obese Women With Polycystic Ovary Syndrome: A Randomized Double-Blind Con-trolled Trial. Reprod. Sci. 2019, 26, 1336–1342, doi:10.1177/1933719118765985.
- Barbieri, R.L. Metformin for the treatment of polycystic ovary syndrome. Obstet. Gynecol. 2003, 101, 785–793, doi:10.1016/s0029-7844(03)00045-0.
- Abu Hashim, H.; Shokeir, T.; Badawy, A. Letrozole versus combined metformin and clomiphene citrate for ovulation in-duction in clomiphene-resistant women with polycystic ovary syndrome: A randomized controlled trial. Fertil. Steril. 2010, 94, 1405–1409, doi:10.1016/j.fertnstert.2009.07.985.
- Bjelica, A.; Trninić-Pjević, A.; Mladenović-Segedi, L.; Cetković, N.; Petrović, D. Comparison of the efficiency of clomiphene citrate and letrozole in combination with metformin in moderately obese clomiphene citrate-resistant polycystic ovarian syndrome patients. Srp. Arh. Celok. Lek. 2016, 144, 146–150, doi:10.2298/SARH1604146B.
- Nemati, M.; Nemati, S.; Taheri, A.M.; Heidari, B. Comparison of metformin and N-acetyl cysteine, as an adjuvant to clomi-phene citrate, in clomiphene-resistant women with polycystic ovary syndrome. J. Gynecol. Obstet. Hum. Reprod. 2017, 46, 579–585, doi:10.1016/j.jogoh.2017.07.004.
- Yu, Y.; Fang, L.; Zhang, R.; He, J.; Xiong, Y.; Guo, X.; Du, Q.; Huang, Y.; Sun, Y. Comparative effectiveness of 9 ovula-tion-induction therapies in patients with clomiphene citrate-resistant polycystic ovary syndrome: A network meta-analysis. Sci. Rep. 2017, 7, 3812, doi:10.1038/s41598-017-03803-9.
- Sawant, S.; Bhide, P. Fertility Treatment Options for Women with Polycystic Ovary Syndrome. Clin. Med. Insights Reprod. Health 2019, 13, 1179558119890867, doi:10.1177/1179558119890867.
- Maged, A.M.; Elsawah, H.; Abdelhafez, A.; Bakry, A.; Mostafa, W.A. The adjuvant effect of metformin and N-acetylcysteine to clomiphene citrate in induction of ovulation in patients with Polycystic Ovary Syndrome. Gynecol. Endocrinol. 2015, 31, 635–638, doi:10.3109/09513590.2015.1037269.
- Abu Hashim, H.; Shokeir, T.; Badawy, A. Letrozole versus combined metformin and clomiphene citrate for ovulation induction in clomiphene-resistant women with polycystic ovary syndrome: A randomized controlled trial. Fertil. Steril. 2010, 94, 1405–1409, doi:10.1016/j.fertnstert.2009.07.985.
- Nemati, M.; Nemati, S.; Taheri, A.M.; Heidari, B. Comparison of metformin and N-acetyl cysteine, as an adjuvant to clomiphene citrate, in clomiphene-resistant women with polycystic ovary syndrome. J. Gynecol. Obstet. Hum. Reprod. 2017, 46, 579–585, doi:10.1016/j.jogoh.2017.07.004.
- Rezk, M.; Shaheen, A.E.; Saif El-Nasr, I. Clomiphene citrate combined with metformin versus letrozole for induction of ovulation in clomiphene-resistant polycystic ovary syndrome: A randomized clinical trial. Gynecol. Endocrinol. 2018, 34, 298–300, doi:10.1080/09513590.2017.1395838.
- Chang, H.H.; Hsueh, Y.S.; Cheng, Y.W.; Ou, H.T.; Wu, M.H. Association between Polymorphisms of OCT1 and Metabolic Response to Metformin in Women with Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2019, 20, 1720, doi:10.3390/ijms20071720.
- Yu, Y.; Fang, L.; Zhang, R.; He, J.; Xiong, Y.; Guo, X.; Du, Q.; Huang, Y.; Sun, Y. Comparative effectiveness of 9 ovulation-induction therapies in patients with clomiphene citrate-resistant polycystic ovary syndrome: A network meta-analysis. Sci. Rep. 2017, 7, 3812, doi:10.1038/s41598-017-03803-9.
- Hoeger, K.; Davidson, K.; Kochman, L.; Cherry, T.; Kopin, L.; Guzick, D.S. The impact of metformin, oral contraceptives, and lifestyle modification on polycystic ovary syndrome in obese adolescent women in two randomized, place-bo-controlled clinical trials. J. Clin. Endocrinol. Metab. 2008, 93, 4299–4306, doi:10.1210/jc.2008-0461.
- Palomba, S.; Materazzo, C.; Falbo, A.; Orio, F.; La Sala, G.B.; Sultan, C. Metformin, oral contraceptives or both to manage oligo-amenorrhea in adolescents with polycystic ovary syndrome? A clinical review. Gynecol. Endocrinol. 2014, 30, 335–340, doi:10.3109/09513590.2013.876001.
- Stefanaki, C.; Bacopoulou, F.; Kandaraki, E.; Boschiero, D.; Diamandi-Kandarakis, E. Lean Women on Metformin and Oral Contraceptives for Polycystic Ovary Syndrome Demonstrate a Dehydrated Osteosarcopenic Phenotype: A Pilot Study. Nu-trients 2019, 11, 2055, doi:10.3390/nu11092055.
- Douchi, T.; Yamamoto, S.; Oki, T.; Maruta, K.; Kuwahata, R.; Nagata, Y. Serum androgen levels and muscle mass in women with polycystic ovary syndrome. Obstet. Gynecol. 1999, 94, 337–340, doi:10.1016/s0029-7844(99)00311-7.
- Cetrone, M.; Mele, A.; Tricarico, D. Effects of the antidiabetic drugs on the age-related atrophy and sarcopenia associated with diabetes type II. Curr. Diabetes Rev. 2014, 10, 231–237, doi:10.2174/1573399810666140918121022.
- Kim, K.H.; Lee, M.S. FGF21 as a mediator of adaptive responses to stress and metabolic benefits of anti-diabetic drugs. J. Endocrinol. 2015, 226, 1–16, doi:10.1530/JOE-15-0160.
- Gupta, A.; Jelinek, H.F.; Al-Aubaidy, H. Glucagon like peptide-1 and its receptor agonists: Their roles in management of Type 2 diabetes mellitus. Diabetes Metab. Syndr. 2017, 11, 225–230, doi:10.1016/j.dsx.2016.09.003.
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756, doi:10.1016/j.cmet.2018.03.001.
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070, doi:10.1002/dmrr.3070.
- Gillani, S.W.; Moosvi, A.F. Clinical Review: Safety and Efficacy Comparison between Sulfonylureas and Dipeptidyl Pepti-dase-4 Inhibitors as Second-Line Therapies in Type 2 Diabetes Mellitus. Curr. Pharm. Des. 2020, 26, 4315–4322, doi:10.2174/1381612826666200408095310.
- Pani, A.; Gironi, I.; Di Vieste, G.; Mion, E.; Bertuzzi, F.; Pintaudi, B. From Prediabetes to Type 2 Diabetes Mellitus in Women with Polycystic Ovary Syndrome: Lifestyle and Pharmacological Management. Int. J. Endocrinol. 2020, 2020, 6276187, doi:10.1155/2020/6276187.
- Romualdi, D.; Versace, V.; Lanzone, A. What is new in the landscape of insulin-sensitizing agents for polycystic ovary syn-drome treatment. Ther. Adv. Reprod. Health 2020, 14, 2633494120908709, doi:10.1177/2633494120908709.
- Jensterle, M.; Kravos, N.A.; Goričar, K.; Janez, A. Short-term effectiveness of low dose liraglutide in combination with met-formin versus high dose liraglutide alone in treatment of obese PCOS: Randomized trial. BMC Endocr. Disord. 2017, 17, 5, doi:10.1186/s12902-017-0155-9.
- Tao, T.; Wu, P.; Wang, Y.; Liu, W. Comparison of glycemic control and β-cell function in new onset T2DM patients with PCOS of metformin and saxagliptin monotherapy or combination treatment. BMC Endocr. Disord. 2018, 18, 14, doi:10.1186/s12902-018-0243-5.
- Elkind-Hirsch, K.E.; Paterson, M.S.; Seidemann, E.L.; Gutowski, H.C. Short-term therapy with combination dipeptidyl pep-tidase-4 inhibitor saxagliptin/metformin extended release (XR) is superior to saxagliptin or metformin XR monotherapy in prediabetic women with polycystic ovary syndrome: A single-blind, randomized, pilot study. Fertil. Steril. 2017, 107, 253–260, doi:10.1016/j.fertnstert.2016.09.023.
- Lamos, E.M.; Malek, R.; Davis, S.N. GLP-1 receptor agonists in the treatment of polycystic ovary syndrome. Expert. Rev. Clin. Pharmacol. 2017, 10, 401–408, doi:10.1080/17512433.2017.1292125.
- Tzotzas, T.; Karras, S.N.; Katsiki, N. Glucagon-Like Peptide-1 (GLP-1) Receptor Agonists in the Treatment of Obese Women with Polycystic Ovary Syndrome. Curr. Vasc. Pharmacol. 2017, 15, 218–229, doi:10.2174/1570161114666161221115324.
- Cena, H.; Chiovato, L.; Nappi, R.E. Obesity, Polycystic Ovary Syndrome, and Infertility: A New Avenue for GLP-1 Receptor Agonists. J. Clin. Endocrinol. Metab. 2020, 105, e2695–e2709, doi:10.1210/clinem/dgaa285.
- Livadas, S.; Androulakis, I.; Angelopoulos, N.; Lytras, A.; Papagiannopoulos, F.; Kassi, G. Liraglutide administration im-proves hormonal/metabolic profile and reproductive features in women with HAIR-AN syndrome. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 19–0150, doi:10.1530/EDM-19-0150.
- Hoeger, K.; Davidson, K.; Kochman, L.; Cherry, T.; Kopin, L.; Guzick, D.S. The impact of metformin, oral contraceptives, and lifestyle modification on polycystic ovary syndrome in obese adolescent women in two randomized, placebo-controlled clinical trials. J. Clin. Endocrinol. Metab. 2008, 93, 4299–4306, doi:10.1210/jc.2008-0461.
- Nestler, J.E.; Jakubowicz, D.J. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after re-duction of insulin secretion in polycystic ovary syndrome. N. Engl. J. Med. 1996, 335, 617–623, doi:10.1056/NEJM199608293350902.
- Chou, K.H.; von Eye Corleta, H.; Capp, E.; Spritzer, P.M. Clinical, metabolic and endocrine parameters in response to met-formin in obese women with polycystic ovary syndrome: A randomized, double-blind and placebo-controlled trial. Horm. Metab. Res. 2003, 35, 86–91, doi:10.1055/s-2003-39056.
- Allen, H.F.; Mazzoni, C.; Heptulla, R.A.; Murray, M.A.; Miller, N.; Koenigs, L.; Reiter, E.O. Randomized controlled trial evaluating response to metformin versus standard therapy in the treatment of adolescents with polycystic ovary syndrome. J. Pediatr. Endocrinol. Metab. 2005, 18, 761–768, doi:10.1515/jpem.2005.18.8.761.
- Bridger, T.; MacDonald, S.; Baltzer, F.; Rodd, C. Randomized placebo-controlled trial of metformin for adolescents with polycystic ovary syndrome. Arch. Pediatr. Adolesc. Med. 2006, 160, 241–246, doi:10.1001/archpedi.160.3.241.
- Diamanti-Kandarakis, E.; Christakou, C.D.; Kandaraki, E.; Economou, F.N. Metformin: An old medication of new fashion: Evolving new molecular mechanisms and clinical implications in polycystic ovary syndrome. Eur. J. Endocrinol. 2010, 162, 193–212, doi:10.1530/EJE-09-0733.
- Al-Zubeidi, H.; Klein, K.O. Randomized clinical trial evaluating metformin versus oral contraceptive pills in the treatment of adolescents with polycystic ovarian syndrome. J. Pediatr. Endocrinol. Metab. 2015, 28, 853–858, doi:10.1515/jpem-2014-0283.
- Al Khalifah, R.A.; Florez, I.D.; Dennis, B.; Thabane, L.; Bassilious, E. Metformin or Oral Contraceptives for Adolescents With Polycystic Ovarian Syndrome: A Meta-analysis. Pediatrics 2016, 137, e20154089, doi:10.1542/peds.2015-4089.
- Sam, S.; Ehrmann, D.А. Metformin therapy for the reproductive and metabolic consequences of polycystic ovary syndrome. Diabetologia 2017, 60, 1656–1661, doi:10.1007/s00125-017-4306-3.
- Kupreeva, M.; Diane, A.; Lehner, R.; Watts, R.; Ghosh, M.; Proctor, S.; Vine, D. Effect of metformin and flutamide on insulin, lipogenic and androgen-estrogen signaling, and cardiometabolic risk in a PCOS-prone metabolic syndrome rodent model. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E16–E33, doi:10.1152/ajpendo.00018.2018.
- Diri, H.; Bayram, F.; Simsek, Y.; Caliskan, Z.; Kocer, D. Comparison of finasteride, metformin, and finasteride plus metfor-min in PCOS. Acta Endocrinol. 2017, 13, 84–89, doi:10.4183/aeb.2017.84.
- Diamanti-Kandarakis, E.; Christakou, C.D.;
- Kandaraki, E.; Economou, F.N. Metformin: An old medication of new fashion: Evolving new molecular mechanisms and clinical implications in polycystic ovary syndrome. Eur. J. Endocrinol. 2010, 162, 193–212, doi:10.1530/EJE-09-0733.
- Kurzthaler, D.; Hadziomerovic-Pekic, D.; Wildt, L.; Seeber, B.E. Metformin induces a prompt decrease in LH-stimulated testosterone response in women with PCOS independent of its insulin-sensitizing effects. Reprod. Biol. Endocrinol. 2014, 12, 98, doi:10.1186/1477-7827-12-98.
- Barber, T.M.; Wass, J.A.; McCarthy, M.I.; Franks, S. Metabolic characteristics of women with polycystic ovaries and oli-go-amenorrhoea but normal androgen levels: Implications for the management of polycystic ovary syndrome. Clin. Endo-crinol. 2007, 66, 513–517, doi:10.1111/j.1365-2265.2007.02764.x.
- Adams, J.M.; Taylor, A.E.; Crowley, W.F.Jr.; Hall, J.E. Polycystic ovarian morphology with regular ovulatory cycles: In-sights into the pathophysiology of polycystic ovarian syndrome. J. Clin. Endocrinol. Metab. 2004, 89, 4343–4350, doi:10.1210/jc.2003-031600.
- Attia, G.R.; Rainey, W.E.; Carr, B.R. Metformin directly inhibits androgen production in human thecal cells. Fertil. Steril. 2001, 76, 517–524, doi:10.1016/s0015-0282(01)01975-6.
- Mansfield, R.; Galea, R.; Brincat, M.; Hole, D.; Mason, H. Metformin has direct effects on human ovarian steroidogenesis. Fertil. Steril. 2003, 79, 956–962, doi:10.1016/s0015-0282(02)04925-7.
- Tosca, L.; Chabrolle, C.; Uzbekova, S.; Dupont, J. Effects of metformin on bovine granulosa cells steroidogenesis: Possible involvement of adenosine 5’ monophosphate-activated protein kinase (AMPK). Biol. Reprod. 2007, 76, 368–378, doi:10.1095/biolreprod.106.055749.
- Tosca, L.; Froment, P.; Solnais, P.; Ferré, P.; Foufelle, F.; Dupont, J. Adenosine 5′-monophosphate-activated protein kinase regulates progesterone secretion in rat granulosa cells. Endocrinology 2005, 146, 4500–4513, doi:10.1210/en.2005-0301.
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endo-crinol. 2018, 9, 753, doi:10.3389/fendo.2018.00753.
- Tosca, L.; Crochet, S.; Ferré, P.; Foufelle, F.; Tesseraud, S.; Dupont, J. AMP-activated protein kinase activation modulates progesterone secretion in granulosa cells from hen preovulatory follicles. J. Endocrinol. 2006, 190, 85–97, doi:10.1677/joe.1.06828.
- Downs, S.M.; Chen, J. Induction of meiotic maturation in mouse oocytes by adenosine analogs. Mol. Reprod. Dev. 2006, 73, 1159–1168, doi:10.1002/mrd.20439.
- LaRosa, C.; Downs, S.M. Meiotic induction by heat stress in mouse oocytes: Involvement of AMP-activated protein kinase and MAPK family members. Biol. Reprod. 2007, 76, 476–486, doi:10.1095/biolreprod.106.057422.
- Mayes, M.A.; Laforest, M.F.; Guillemette, C.; Gilchrist, R.B.; Richard, F.J. Adenosine 5′-monophosphate kinase-activated protein kinase (PRKA) activators delay meiotic resumption in porcine oocytes. Biol. Reprod. 2007, 76, 589–597, doi:10.1095/biolreprod.106.057828.
- Bilodeau-Goeseels, S. Cows are not mice: The role of cyclic AMP, phosphodiesterases, and adenosine monophosphate-activated protein kinase in the maintenance of meiotic arrest in bovine oocytes. Mol. Reprod. Dev. 2011, 78, 734–743, doi:10.1002/mrd.21337.
- Bertoldo, M.J.; Faure, M.; Dupont, J.; Froment, P. AMPK: A master energy regulator for gonadal function. Front. Neurosci. 2015, 9, 235, doi:10.3389/fnins.2015.00235.
- Reverchon, M.; Cornuau, M.; Cloix, L.; Ramé, C.; Guerif, F.; Royère, D.; Dupont, J. Visfatin is expressed in human granulosa cells: Regulation by metformin through AMPK/SIRT1 pathways and its role in steroidogenesis. Mol. Hum. Reprod. 2013, 19, 313–326, doi:10.1093/molehr/gat002.
- Bertoldo, M.J.; Guibert, E.; Faure, M.; Ramé, C.; Foretz, M.; Viollet, B.; Dupont, J.; Froment, P. Specific deletion of AMP-activated protein kinase (α1AMPK) in murine oocytes alters junctional protein expression and mitochondrial physi-ology. PLoS ONE 2015, 10, e0119680, doi:10.1371/journal.pone.0119680.
- Tosca, L.; Solnais, P.; Ferré, P.; Foufelle, F.; Dupont, J. Metformin-induced stimulation of adenosine 5’ monophosphate-activated protein kinase (PRKA) impairs progesterone secretion in rat granulosa cells. Biol. Reprod. 2006, 75, 342–351, doi:10.1095/biolreprod.106.050831
- Tosca, L.; Ramé, C.; Chabrolle, C.; Tesseraud, S.; Dupont, J. Metformin decreases IGF1-induced cell proliferation and protein synthesis through AMP-activated protein kinase in cultured bovine granulosa cells. Reproduction 2010, 139, 409–418, doi:10.1530/REP-09-0351.
- Xu, Y.; Gao, Y.; Huang, Z.; Zheng, Y.; Teng, W.; Zheng, D.; Zheng, X. LKB1 suppresses androgen synthesis in a mouse model of hyperandrogenism via IGF-1 signaling. FEBS Open Bio 2019, 9, 1817–1825, doi:10.1002/2211-5463.12723.
- Huhtala, M.S.; Tertti, K.; Juhila, J.; Sorsa, T.; Rönnemaa, T. Metformin and insulin treatment of gestational diabetes: Effects on inflammatory markers and IGF-binding protein-1—Secondary analysis of a randomized controlled trial. BMC Pregnancy Childbirth 2020, 20, 401, doi:10.1186/s12884-020-03077-6.
- Nestler, J.E.; Powers, L.P.; Matt, D.W.; Steingold, K.A.; Plymate, S.R.; Rittmaster, R.S.; Clore, J.N.; Blackard, W.G. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syn-drome. J. Clin. Endocrinol. Metab. 1991, 72, 83–89, doi:10.1210/jcem-72-1-83.
- Botwood, N.; Hamilton-Fairley, D.; Kiddy, D.; Robinson, S.; Franks, S. Sex hormone-binding globulin and female reproduc-tive function. J. Steroid Biochem. Mol. Biol. 1995, 53, 529–531, doi:10.1016/0960-0760(95)00108-c.
- Franks, S.; Kiddy, D.S.; Hamilton-Fairley, D.; Bush, A.; Sharp, P.S.; Reed, M.J. The role of nutrition and insulin in the regula-tion of sex hormone binding globulin. J. Steroid Biochem. Mol. Biol. 1991, 39, 835–838, doi:10.1016/0960-0760(91)90033-2.
- Jiang, Z.Z.; Hu, M.W.; Ma, X.S.; Schatten, H.; Fan, H.Y.; Wang, Z.B.; Sun, Q.Y. LKB1 acts as a critical gatekeeper of ovarian primordial follicle pool. Oncotarget 2016, 7, 5738–5753, doi:10.18632/oncotarget.6792.
- Morris, D.V.; Falcone, T. The relationship between insulin sensitivity and insulin-like growth factor-binding protein-1. Gy-necol. Endocrinol. 1996, 10, 407–412, doi:10.3109/09513599609023605.
- Landay, M.; Huang, A.; Azziz, R. Degree of hyperinsulinemia, independent of androgen levels, is an important determinant of the severity of hirsutism in PCOS. Fertil. Steril. 2009, 92, 643–647, doi:10.1016/j.fertnstert.2008.06.021.
- Firmansyah, A.; Chalid, M.T.; Farid, R.B.; Nusratuddin, N. The correlation between insulin-like growth factor binding pro-tein 1 (IGFBP-1) and homeostasis model assessment of insulin resistance (HOMA-IR) in polycystic ovarian syndrome with insulin resistance. Int. J. Reprod. Biomed. 2018, 16, 679–682.
- Barbieri, R.L. Hyperandrogenism, insulin resistance and acanthosis nigricans. 10 years of progress. J. Reprod. Med. 1994, 39, 327–336.
- Kelly, C.J.; Stenton, S.R.; Lashen, H. Insulin-like growth factor binding protein-1 in PCOS: A systematic review and me-ta-analysis. Hum. Reprod. Update 2011, 17, 4–16, doi:10.1093/humupd/dmq027.
- Bergh, C.; Carlsson, B.; Olsson, J.H.; Selleskog, U.; Hillensjö, T. Regulation of androgen production in cultured human thecal cells by insulin-like growth factor I and insulin. Fertil. Steril. 1993, 59, 323–331, doi:10.1016/s0015-0282(16)55675-1.
- Mason, H.D.; Margara, R.; Winston, R.M.; Seppala, M.; Koistinen, R.; Franks, S. Insulin-like growth factor-I (IGF-I) inhibits production of IGF-binding protein-1 while stimulating estradiol secretion in granulosa cells from normal and polycystic human ovaries. J. Clin. Endocrinol. Metab. 1993, 76, 1275–1279, doi:10.1210/jcem.76.5.7684393.
- Cibula, D.; Fanta, M.; Vrbikova, J.; Stanicka, S.; Dvorakova, K.; Hill, M.; Skrha, J.; Zivny, J.; Skrenkova, J. The effect of com-bination therapy with metformin and combined oral contraceptives (COC) versus COC alone on insulin sensitivity, hy-perandrogenaemia, SHBG and lipids in PCOS patients. Hum. Reprod. 2005, 20, 180–184, doi:10.1093/humrep/deh588.
- Wei, W.; Zhao, H.; Wang, A.; Sui, M.; Liang, K.; Deng, H.; Ma, Y.; Zhang, Y.; Zhang, H.; Guan, Y. A clinical study on the short-term effect of berberine in comparison to metformin on the metabolic characteristics of women with polycystic ovary syndrome. Eur. J. Endocrinol. 2012, 166, 99–105, doi:10.1530/EJE-11-0616.
- Pasquali, R.; Gambineri, A.; Biscotti, D.; Vicennati, V.; Gagliardi, L.; Colitta, D.; Fiorini, S.; Cognigni, G.E.; Filicori, M.; Morselli-Labate, A.M. Effect of long-term treatment with metformin added to hypocaloric diet on body composition, fat distribution, and androgen and insulin levels in abdominally obese women with and without the polycystic ovary syn-drome. J. Clin. Endocrinol. Metab. 2000, 85, 2767–2774, doi:10.1210/jcem.85.8.6738.
- Wassell, J.; Michail, M.; Soliman, N.; Wardle, P.G. The value of sex hormone binding globulin (SHBG) in predicting treat-ment response in polycystic ovary syndrome (PCOS). Clin. Lab. 2011, 57, 95–98.
- Zhang, J.; Si, Q.; Li, J. Therapeutic effects of metformin and clomiphene in combination with lifestyle intervention on infer-tility in women with obese polycystic ovary syndrome. Pak. J. Med. Sci. 2017, 33, 8–12, doi:10.12669/pjms.331.11764.
- Furat Rencber, S.; Kurnaz Ozbek, S.; Eraldemır, C.; Sezer, Z.; Kum, T.; Ceylan, S.; Guzel, E. Effect of resveratrol and metfor-min on ovarian reserve and ultrastructure in PCOS: An experimental study. J. Ovarian Res. 2018, 11, 55, doi:10.1186/s13048-018-0427-7.
- Kadoura, S.; Alhalabi, M.; Nattouf, A.H. Effect of Calcium and Vitamin D Supplements as an Adjuvant Therapy to Metfor-min on Menstrual Cycle Abnormalities, Hormonal Profile, and IGF-1 System in Polycystic Ovary Syndrome Patients: A Randomized, Placebo-Controlled Clinical Trial. Adv. Pharmacol. Sci. 2019, 2019, 9680390, doi:10.1155/2019/9680390.
- Song, Y.; Wang, H.; Huang, H.; Zhu, Z. Comparison of the efficacy between NAC and metformin in treating PCOS patients: A meta-analysis. Gynecol. Endocrinol. 2020, 36, 204–210, doi:10.1080/09513590.2019.1689553.
- Morales, A.J.; Laughlin, G.A.; Bützow, T.; Maheshwari, H.; Baumann, G.; Yen, S.S. Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: Common and distinct features. J. Clin. Endocrinol. Metab. 1996, 81, 2854–2864, doi:10.1210/jcem.81.8.8768842.
- Chang, R.J. The reproductive phenotype in polycystic ovary syndrome. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 688–695, doi:10.1038/ncpendmet0637.
- Sinha, P.; Chitra, T.; Papa, D.; Nandeesha, H. Laparoscopic Ovarian Drilling Reduces Testosterone and Luteinizing Hor-mone/Follicle-Stimulating Hormone Ratio and Improves Clinical Outcome in Women with Polycystic Ovary Syndrome. J. Hum. Reprod. Sci. 2019, 12, 224–228, doi:10.4103/jhrs.JHRS_161_18.
- Dulka, E.A.; Burger, L.L.; Moenter, S.M. Ovarian Androgens Maintain High GnRH Neuron Firing Rate in Adult Prenatal-ly-Androgenized Female Mice. Endocrinology 2020, 161, bqz038, doi:10.1210/endocr/bqz038.
- McCartney, C.R.; Campbell, R.E. Abnormal GnRH Pulsatility in Polycystic Ovary Syndrome: Recent Insights. Curr. Opin. Endocr. Metab. Res. 2020, 12, 78–84, doi:10.1016/j.coemr.2020.04.005.
- Woo, I.; Tobler, K.; Khafagy, A.; Christianson, M.S.; Yates, M.; Garcia, J. Predictive Value of Elevated LH/FSH Ratio for Ov-ulation Induction in Patients with Polycystic Ovary Syndrome. J. Reprod. Med. 2015, 60, 495–500.
- Roland, A.V.; Moenter, S.M. Prenatal androgenization of female mice programs an increase in firing activity of gonadotro-pin-releasing hormone (GnRH) neurons that is reversed by metformin treatment in adulthood. Endocrinology 2011, 152, 618–628, doi:10.1210/en.2010-0823.
- Maciel, G.A.; Hayashida, S.A.; da Costa, L.C.; Marcondes, J.A.; da Fonseca, A.M.; Soares, J.M.Jr.; Baracat, E.C. Influence of LH and high-density lipoprotein cholesterol (HDL-C) on metformin response in women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 157, 180–184, doi:10.1016/j.ejogrb.2011.03.028.
- Lv, W.S.; Wen, J.P.; Li, L.; Sun, R.X.; Wang, J.; Xian, Y.X.; Cao, C.X.; Wang, Y.L.; Gao, Y.Y. The effect of metformin on food intake and its potential role in hypothalamic regulation in obese diabetic rats. Brain Res. 2012, 1444, 11–19, doi:10.1016/j.brainres.2012.01.028.
- McIlwraith, E.K.; Belsham, D.D. Hypothalamic reproductive neurons communicate through signal transduction to control reproduction. Mol. Cell. Endocrinol. 2020, 110971, doi:10.1016/j.mce.2020.110971.
- Malin, S.K.; Kashyap, S.R. Effects of metformin on weight loss: Potential mechanisms. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 323–329, doi:10.1097/MED.0000000000000095.
- Derkach, K.V.; Zakharova, I.O.; Romanova, I.V.; Zorina, I.I.; Mikhrina, A.L.; Shpakov, A.O. Metabolic parameters and func-tional state of hypothalamic signaling systems in AY/a mice with genetic predisposition to obesity and the effect of met-formin. Dokl. Biochem. Biophys. 2017, 477, 377–381, doi:10.1134/S1607672917060096.
- Derkach, K.V.; Zakharova, I.O.; Zorina, I.I.; Bakhtyukov, A.A.; Romanova, I.V.; Bayunova, L.V.; Shpakov, A.O. The evidence of metabolic-improving effect of metformin in Ay/a mice with genetically-induced melanocortin obesity and the contribu-tion of hypothalamic mechanisms to this effect. PLoS ONE 2019, 14, e0213779, doi:10.1371/journal.pone.0213779.
- Yerevanian, A.; Soukas, A.A. Metformin: Mechanisms in Human Obesity and Weight Loss. Curr. Obes. Rep. 2019, 8, 156–164, doi:10.1007/s13679-019-00335-3.
- Hausman, G.J.; Barb, C.R.; Lents, C.A. Leptin and reproductive function. Biochimie 2012, 94, 2075–2081, doi:10.1016/j.biochi.2012.02.022.
- Zhang, J.; Gong, M. Review of the role of leptin in the regulation of male reproductive function. Andrologia 2018, doi:10.1111/and.12965.
- Huang, Y.; Yu, Y.; Gao, J.; Li, R.; Zhang, C.; Zhao, H.; Zhao, Y.; Qiao, J. Impaired oocyte quality induced by dehydroepi-androsterone is partially rescued by metformin treatment. PLoS ONE 2015, 10, e0122370, doi:10.1371/journal.pone.0122370.
- Jin, J.; Ma, Y.; Tong, X.; Yang, W.; Dai, Y.; Pan, Y.; Ren, P.; Liu, L.; Fan, H.Y.; Zhang, Y.; et al. Metformin inhibits testos-terone-induced endoplasmic reticulum stress in ovarian granulosa cells via inactivation of p38 MAPK. Hum. Reprod. 2020, 35, 1145–1158, doi:10.1093/humrep/deaa077.
- Harada, M.; Nose, E.; Takahashi, N.; Hirota, Y.; Hirata, T.; Yoshino, O.; Koga, K.; Fujii, T.; Osuga, Y. Evidence of the activa-tion of unfolded protein response in granulosa and cumulus cells during follicular growth and maturation. Gynecol. Endo-crinol. 2015, 31, 783–787, doi:10.3109/09513590.2015.1062862.
- Park, H.J.; Park, J.Y.; Kim, J.W.; Yang, S.G.; Jung, J.M.; Kim, M.J.; Kang, M.J.; Cho, Y.H.; Wee, G.; Yang, H.Y.; et al. Melatonin improves the meiotic maturation of porcine oocytes by reducing endoplasmic reticulum stress during in vitro maturation. J. Pineal Res. 2018, 64, e12458, doi:10.1111/jpi.12458.
- Tobiume, K.; Matsuzawa, A.; Takahashi, T.; Nishitoh, H.; Morita, K.; Takeda, K.; Minowa, O.; Miyazono, K.; Noda, T.; Ichijo, H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2, 222–228, doi:10.1093/embo-reports/kve046.
- Kim, H.S.; Kim, Y.; Lim, M.J.; Park, Y.G.; Park, S.I.; Sohn, J. The p38-activated ER stress-ATF6α axis mediates cellular senes-cence. FASEB J. 2019, 33, 2422–2434, doi:10.1096/fj.201800836R.
- Azhary, J.M.K.; Harada, M.; Takahashi, N.; Nose, E.; Kunitomi, C.; Koike, H.; Hirata, T.; Hirota, Y.; Koga, K.; Wada-Hiraike, O.; et al. Endoplasmic Reticulum Stress Activated by Androgen Enhances Apoptosis of Granulosa Cells via Induction of Death Receptor 5 in PCOS. Endocrinology 2019, 160, 119–132, doi:10.1210/en.2018-00675.
- Rice, S.; Pellatt, L.; Ramanathan, K.; Whitehead, S.A.; Mason, H.D. Metformin inhibits aromatase via an extracellular sig-nal-regulated kinase-mediated pathway. Endocrinology 2009, 150, 4794–4801, doi:10.1210/en.2009-0540.
- Rice, S.; Elia, A.; Jawad, Z.; Pellatt, L.; Mason, H.D. Metformin inhibits follicle-stimulating hormone (FSH) action in human granulosa cells: Relevance to polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 1491–1500, doi:10.1210/jc.2013-1865.
- Fuhrmeister, I.P.; Branchini, G.; Pimentel, A.M.; Ferreira, G.D.; Capp, E.; Brum, I.S.; von Eye Corleta, H. Human granulosa cells: Insulin and insulin-like growth factor-1 receptors and aromatase expression modulation by metformin. Gynecol. Ob-stet. Invest. 2014, 77, 156–162, doi:10.1159/000358829.
- Catteau-Jonard, S.; Jamin, S.P.; Leclerc, A.; Gonzalès, J.; Dewailly, D.; di Clemente, N. Anti-Mullerian hormone, its receptor, FSH receptor, and androgen receptor genes are overexpressed by granulosa cells from stimulated follicles in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 4456–4461, doi:10.1210/jc.2008-1231.
- González-Fernández, R.; Peña, Ó.; Hernández, J.; Martín-Vasallo, P.; Palumbo, A.; Ávila, J. Patients with endometriosis and patients with poor ovarian reserve have abnormal follicle-stimulating hormone receptor signaling pathways. Fertil. Steril. 2011, 95, 2373–2378, doi:10.1016/j.fertnstert.2011.03.030.
- Dewailly, D.; Robin, G.; Peigne, M.; Decanter, C.; Pigny, P.; Catteau-Jonard, S. Interactions between androgens, FSH, an-ti-Müllerian hormone and estradiol during folliculogenesis in the human normal and polycystic ovary. Hum. Reprod. Update 2016, 22, 709–724, doi:10.1093/humupd/dmw027.
- Geng, Y.; Sui, C.; Xun, Y.; Lai, Q.; Jin, L. MiRNA-99a can regulate proliferation and apoptosis of human granulosa cells via targeting IGF-1R in polycystic ovary syndrome. J. Assist. Reprod. Genet. 2019, 36, 211–221, doi:10.1007/s10815-018-1335-x.
- He, T.; Liu, Y.; Zhao, S.; Liu, H.; Wang, Z.; Shi, Y. Comprehensive assessment the expression of core elements related to IG-FIR/PI3K pathway in granulosa cells of women with polycystic ovary syndrome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 233, 134–140, doi:10.1016/j.ejogrb.2018.12.010.
- He, T.; Sun, Y.; Zhang, Y.; Zhao, S.; Zheng, Y.; Hao, G.; Shi, Y. MicroRNA-200b and microRNA-200c are up-regulated in PCOS granulosa cell and inhibit KGN cell proliferation via targeting PTEN. Reprod. Biol. Endocrinol. 2019, 17, 68, doi:10.1186/s12958-019-0505-8.
- Mason, H.D.; Willis, D.S.; Holly, J.M.; Franks, S. Insulin preincubation enhances insulin-like growth factor-II (IGF-II) action on steroidogenesis in human granulosa cells. J. Clin. Endocrinol. Metab. 1994, 78, 1265–1267, doi:10.1210/jcem.78.5.8175988.
- Willis, D.S.; Mason, H.D.; Watson, H.; Franks, S. Developmentally regulated responses of human granulosa cells to insu-lin-like growth factors (IGFs): IGF-I and IGF-II action mediated via the type-I IGF receptor. J. Clin. Endocrinol. Metab. 1998, 83, 1256–1259, doi:10.1210/jcem.83.4.4699.
- Palomba, S.; Falbo, A.; Orio, F., Jr.; Manguso, F.; Russo, T.; Tolino, A.; Annamaria, C.; Dale, B.; Zullo, F. A randomized con-trolled trial evaluating metformin pre-treatment and co-administration in non-obese insulin-resistant women with poly-cystic ovary syndrome treated with controlled ovarian stimulation plus timed intercourse or intrauterine insemination. Hum. Reprod. 2005, 20, 2879–2886, doi:10.1093/humrep/dei130.
- Huang, X.; Wang, P.; Tal, R.; Lv, F.; Li, Y.; Zhang, X. A systematic review and meta-analysis of metformin among patients with polycystic ovary syndrome undergoing assisted reproductive technology procedures. Int. J. Gynaecol. Obstet. 2015, 131, 111–116, doi:10.1016/j.ijgo.2015.04.046.
- Wu, L.L.; Russell, D.L.; Norman, R.J.; Robker, R.L. Endoplasmic reticulum (ER) stress in cumulus-oocyte complexes impairs pentraxin-3 secretion, mitochondrial membrane potential (DeltaPsi m), and embryo development. Mol. Endocrinol. 2012, 26, 562–573, doi:10.1210/me.2011-1362.