Immune checkpoint inhibitors (ICIs) improve the survival of patients with multiple types of cancer. However, low response rates and atypical responses limit their success in clinical applications. The paradoxical acceleration of tumor growth after treatment, defined as hyperprogressive disease (HPD), is the most difficult problem facing clinicians and patients alike. The mechanisms that underlie hyperprogression (HP) are still unclear and controversial, although a large number of studies have investigated the phenomenon and several associated factors have been reported. Gamma-interferon (IFN-γ) is a key cytokine in antitumor response and its levels increase during ICI therapy. Even though this factor has been widely associated with resistance to ICI therapy, it has not yet been demonstrated to be directly associated with HP. Nevertheless, data suggest that IFN-γ may contribute to HP onset through different mechanisms, including the activation of the inflammasome pathway, the expression of the immunosuppressive enzyme IDO1 and the triggering of activation-induced cell death (AICD) in effector T cells. These findings make IFN-γ worthy of attention in the context of HPD development.
- hyperprogression
- hyperprogressive disease
- cancer
- immune checkpoint inhibitors
- immunotherapy
- IFN-γ
- tumor microenvironment
- MDSC
- IDO1
- AICD
1. Introduction
Cancer immunotherapy aims to strengthen the immune system against tumors. The introduction of immunotherapy into clinical practice has provided clinicians with an innovative tool for the treatment of various solid and hematologic malignancies[1]. One of the most successful strategies is the administration of immune checkpoint inhibitors (ICIs), which are a wide range of monoclonal antibodies directed toward immune checkpoint (IC) proteins that are expressed on tumor cell and/or immune cell surfaces. The targeting of ICs reverses tumor-mediated immunosuppression and awakens immune responses[2]. The use of ICIs, either as monotherapy or combo-therapy, has shown favorable outcomes and remarkably long-term responses in patients with a large variety of cancer types, especially malignant melanoma and lung cancer[3][4][5]. The Food and Drug Administration (FDA) has so far approved seven ICIs that target cytotoxic T-lymphocyte antigen 4 (CTLA-4), programmed death-1 (PD-1) and PD-ligand 1 (PD-L1) for the treatment of several tumor types[5]. Nevertheless, response rates, according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, for IC blockade in patients with solid tumors, range from 18 to 40% [6][7], as the beneficial clinical effects of ICI therapy are not long-lasting in some cases [8]. Moreover, the unique mechanism of action of ICIs can lead to unconventional responses, making IC blockade harmful to a subset of patients. Among these novel responses, the most relevant, in terms of negative clinical outcome, is hyperprogressive disease (HPD), a paradoxical acceleration of tumor growth induced by ICI therapy[8]. Even though several factors have been proposed as leading causes of HPD development, including alterations in T-cell subpopulations [9][10][11][12], tumor cells[13][14][15][16][17][18][19][20], cytokine secretion[21][22][23][24] and inflammation[25][26], the mechanisms underlying hyperprogression (HP) after ICI therapy remain unknown.
2. Role of IFN-γ in HPD development
Gamma-interferon (IFN-γ) is a major regulatory and effector cytokine predominantly produced by T and natural killer (NK) cells in response to inflammatory and immune stimuli. In the tumor microenvironment (TME), IFN-γ, mainly produced by infiltrating T lymphocytes (TILs), is a key player in tumor immunosurveillance. The antitumor action includes antiproliferative, antiangiogenic and proapoptotic effects[27][28][29][30][31], in addition to the upregulation of major histocompatibility complex (MHC) class I molecules on tumor cells[32][33]. Moreover, IFN-γ activates CD8+ cytotoxic T lymphocytes, CD4+ Th1 cells, NK cells, dendritic cells (DCs) and macrophages, and stimulates the latter to switch towards the tumoricidal and proinflammatory M1 phenotype[33][34][35]. Conversely, IFN-γ also inhibits regulatory T (Treg) cell differentiation and function[36].
On the other hand, IFN-γ exerts a paradoxical immunosuppressive role that supports tumor progression and dissemination[37][38][39]. For instance, the activation of the IFN-γ receptor (IFNGR) on tumor cells activates the JAK/STAT signaling pathway, resulting in PD-L1 upregulation[40][41][42]. Nevertheless, alterations in the JAK/STAT pathway have been frequently associated with resistance to ICI therapy[43][44][45]]. Human melanoma cell lines with loss-of-function mutations in either JAK1 or JAK2 do not express IFN-γ-response genes after IFN-γ exposure. The analysis of the transcriptome of advanced melanoma under ICI therapy has highlighted an association between clinical response to treatment and the expression of IFN-γ-response genes involved in MHC class I and II upregulation[46]. Resistance to ICI therapy may therefore be due both to the incapacity of tumor cells to induce the full set of IFN-γ-response genes and to the loss of sensitivity to IFN-γ signaling. Interestingly, prolonged IFN-γ receptor signaling in tumor cells can also mediate resistance to ICIs through epigenomic changes in the JAK/STAT pathway[47]. IFN-γ can even support tumorigenesis by influencing the TME. The IFN-γ signaling pathway can indeed increase angiogenesis in the TME by inhibiting the expression of vascular endothelial growth inhibitor (VEGI) [48]. IFN-γ also suppresses the action of immune effector cells via the upregulation of immunosuppressive cytokines, including IL-21, IL-27 and IL-35[49][50][51][52][53][54], and by the recruitment and differentiation of Treg cells and myeloid-derived suppressor cells (MDSCs)[55][56][57][58][59][60][61][62][63].
All of the above-reported examples highlight the role that IFN-γ plays in tumor resistance to ICI therapy. Based on this evidence, it is reasonable to assume that IFN-γ is worthy of investigation in the context of HPD.
2.1. IFN-γ and Inflammasome
Several studies reported a negative correlation between MDSCs and the response to ICIs[64][65][66], leading to the suggestion that MDCSs can be a negative predictive marker for ICI therapy[67]. A few case reports noted a correlation between HPD development and the number of MDSCs in the peripheral blood and in the TME of patients[68][69]. Moreover, the recruitment of granulocytic MDSCs to the TME following ICI therapy, through the IFN-γ-dependent activation of the inflammasome pathway in cancer cells, has been reported[70]. PD-L1 upregulation by IFN-γ after ICIs and the consequent activation of the PD-L1 intrinsic signaling pathway in tumor cells trigger the activation of NLR family pyrin domain containing 3 (NLRP3), which leads to the downstream activation of the heat-shock proteins 70 (HSP70)/Toll-like receptor 4 (TLR4) signaling pathway and Wnt5a production. This signaling cascade ultimately leads to C-X-C motif chemokine ligand 5 (CXCL5) release, resulting in chemokine-dependent recruitment of polymorphonuclear-like MDSCs[70]. PD-L1 triggers NLRP3 activation by repressing STAT3, which is a transcription factor involved in IFN-cytotoxicity. Mutations in the intracytoplasmic DTSSK domain of PD-L1, which is a conserved sequence that acts as a negative regulator of PD-L1 functions, lead to hyperactive PD-L1 molecules in human tumors, enhancing the capacity of the PD-L1 intracellular pathway to interfere with STAT3 expression and phosphorylation[19]. Thus, in patients carrying mutations in the intracytoplasmic domain of PD-L1, the molecules of the inflammasome may be further augmented by PD-L1 upregulation after IFN-γ secretion in response to ICIs, eventually leading to HPD. Interestingly, a mutational analysis performed on tumors after pembrolizumab treatment highlighted the presence of missense or indel mutations in genes involved in the negative regulation of NLRP3 activation and inflammasome pathway[69], including the caspase recruitment domain (CARD8 and CARD11), protein flightless-1 homolog (FLII) and nuclear factor erythroid 2-related factor 2 (NFE2L2)[71][72][73][74]. Moreover, hyperprogressive tumors show mutations in NOTCH1, which seems to be involved in NLRP3 activation and inflammasome pathway [13][69][75][76][77]. In addition, the mechanism of MDSC recruitment in HPD may also be related to the impairment of effector T-cell activity, resulting in the expansion of Treg cells, the inhibition of NK cells and the secretion of immunosuppressive cytokines[78]. On the basis of the above-reported evidence, it can therefore be stated that IFN-γ-mediated recruitment of MDSCs in the TME may be a relevant aspect of HPD development.
2.2. IFN-γ and IDO1
Paracrine Wnt5a signaling is also involved in DC upregulation and enzymatic activity of indoleamine 2,3-dioxygenase (IDO1), promoting DC-mediated Treg differentiation[79][80]. IDO1 is a cytosolic enzyme that contributes to immune regulation by inducing metabolic changes in the local microenvironment. The enzyme catalyzes the rate-limiting step of tryptophan metabolism, which converts tryptophan (trp) into the downstream catabolite kynurenine (kyn). IDO1 is physiologically expressed by professional antigen-presenting cells (APCs), as well as by epithelial cells, the vascular endothelium and peripheral lymphoid organs, and acts as a peripheral IC, contributing to host defense against infection, peripheral immune tolerance, inhibition of local inflammation and autoimmunity [81][82]. Tumor cells use IDO1 expression as a mechanism of immune escape[83][84][85]. Trp depletion indeed results in a blockade of T-cell protein synthesis[86][87], while kyn and its derivatives induce PD-1 expression on activated T cells, together with the differentiation of Treg cells and tolerogenic DCs through aryl hydrocarbon receptor (AhR) activation[88][89][90][91]. In addition, IDO1 expression is induced, as is that of PD-L1, when some degree of inflammation occurs in the tumor, e.g., the presence of proinflammatory mediators, such as IFN-γ, as a mechanism of adaptive resistance against infiltrating T cells[92][93][94]. This aspect clearly has negative implications for ICI therapy, since increased IDO1 activity may increase tumor-infiltrating Treg cells, decrease TILs and accelerate tumor growth[95][96][97][98][99]. In non-small-cell lung cancer (NSCLC) patients treated with nivolumab, serum kyn/trp ratio was higher in early progressors with intrinsic resistance to anti-PD-1 therapy. Moreover, patients with high kyn/trp ratios showed a progression-free survival of three months, which is very similar to that of hyperprogressive patients in a study by Champiat et al. [100][101]. This evidence suggests that IDO1 induction by IFN-γ after ICI therapy may counteract the effectiveness of an otherwise beneficial treatment. Combination treatment with ICIs and IDO1 inhibitors in preclinical studies has been observed to enhance the infiltration and proliferation of effector T cells in the TME[102][103]. However, despite encouraging clinical results in early phase trials[104][105][106], in a randomized phase III study, patients with metastatic melanoma treated with both the IDO1 inhibitor epacadostat and pembrolizumab, had no benefit from the combined therapy, in comparison to pembrolizumab monotherapy[107]. This negative result may, however, be caused by limited preclinical data and either the incomplete inhibition of IDO1 or the compensatory expression of other trp-degrading enzymes[108]. Further clinical studies are therefore needed to understand whether this combined therapy may have therapeutic potential.
Interestingly, IDO1 upregulation is inversely correlated with p53[109], whose expression can be suppressed by IDO1 via the c-Jun N-terminal kinase (JNK) pathway[110]. It is notable that the downregulation of p53 has already been suggested as a HPD mechanism in patients with mouse double minute homolog 2 (MDM2) amplification[13]. Moreover, IDO1 expression is modulated by transforming growth factor-beta (TGF-β) via the Fyn-dependent phosphorylation of immunoreceptor tyrosine-based inhibition motifs (ITIMs) in IDO1, and the activation of the NF-κB pathway, which lead to a tolerogenic phenotype in DCs[111]. TGF-β can also activate the JNK pathway through TGF-β activated kinase 1 (TAK1) and c-Jun phosphorylation[112][113]. It is interesting to note that the TGF-β signaling pathway has been found to be transcriptionally upregulated in HPD tumors following IC blockade, as compared to treatment-naive tumors[69]. Therefore, HPD patients with IDO1-expressing tumors may present a hyperactivated JNK pathway which may result in p53 suppression. Moreover, post-therapy HPD tumors have also displayed transcriptional upregulation of PI3K/AKT and MAPK/ERK pathways, and it has been shown that PI3K and MAPK oncogenic mutations can favor constitutive IDO1 expression on tumor cells[69][114][115].
2.3. IFN-γ and Activation-Induced Cell Death
A further hypothesis that involves IFN-γ in HPD development, focuses on the differential immunological actions of IC blockade that occur depending on tumor burden. The combination of anti-CTLA-4 and anti-PD-1 therapy in mice with high tumor burden (HTB) leads to improved tumor control and to the generation of more activated antigen-specific T cells, as compared to mice with low tumor burden (LTB), in which combination treatment has been shown to compromise antitumor immune response, inducing the loss of antigen-specific T cells[116]. This finding was supported by retrospective clinical data from metastatic melanoma patients who received either monotherapy or combination therapy. Patients treated with dual IC blockade showed significantly lower response rates than those treated with monotherapy in low disease settings, but not in higher disease settings. The detrimental effect of combined therapy in the LTB state was associated with higher IFN-γ production, which was responsible for tumor-reactive CD8+ T-cell apoptosis via activation-induced cell death (AICD). AICD physiologically takes place in the early CD4+ and CD8+ T-cell priming stage, and leads to cell apoptosis to prevent immune hyperactivation. IFN-γ signaling is the key factor in activating this process, together with IL-2[117]. The induction of IFN-γ secretion after dual-blockade treatments can promote the apoptosis of tumor-reactive CD8+ T cells in the LTB setting, limiting the formation of effector memory antitumor responses. In the HTB state, prolonged antigen exposure may lead to T cells with a markedly exhausted phenotype, which may be more prone to reinvigoration after IC blockade. By contrast, in LTB or in the early tumor setting, short-term antigen exposure may be unable to induce a fully exhausted phenotype in T cells. Therefore, the activation of T-cell-receptor signaling against tumor antigens, in combination with dual IC blockade, may result in immune hyperactivation, triggering the AICD process. These findings appear to indicate that the paradoxical effect of IFN-γ in tumor response might derive from the differential exhaustion status of T cells in response to ICIs. Although the mechanisms underlying AICD have not yet been fully understood, Fas seems to be the major death receptor responsible for triggering the AICD pathway in CD4+ T cells[118]. Moreover, STAT1 and caspase 8, which are activated by the IFN-γ pathway, may be involved in the process[117]. The possible role of the activation of the AICD pathway by IFN-γ in HPD is supported by a study in which two hyperprogressive patients displayed depletion of the immune-cell populations involved in tumor clearance, including monocytes, central memory CD4+ T cells, NK cells and activated DCs[69]. In these patients, anti-PD-1 therapy had probably induced an accelerated AICD process in the antitumor activating lymphocytes, as suggested by the activation of apoptosis gene sets and the upregulation of marker genes in the bcl-2 pathway after treatment. These studies suggest that, in some patients, ICI therapy may be responsible for excessive activation of the immune response, which could trigger regulatory mechanisms and hinder therapeutic antitumor effects.
In conclusion, it may be suggested that IFN-γ contributes to HPD onset in predisposed patients via the induction of the inflammasome pathway and consequent MDSC recruitment, the induction of IDO1 activity, which may result in the downregulation of p53 in tumor cells, and, finally, the activation of AICD, which leads to T-cell depletion (Figure 1).
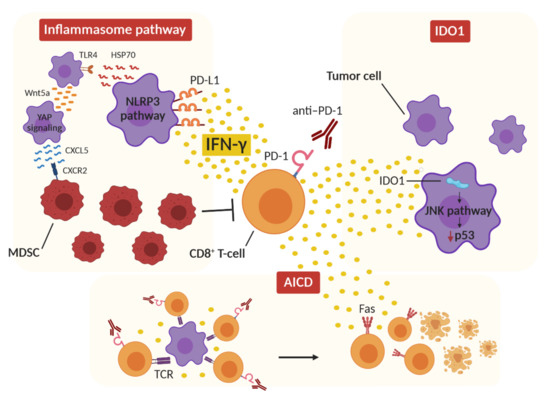
Figure 1. Proposed IFN-γ-dependent mechanisms of hyperprogression. The release of IFN-γ from CD8+ T cells after ICI therapy can activate the inflammasome pathway by upregulating PD-L1 expression on tumor cells and activating NLRP3 signaling, resulting in immunosuppressive MDSC recruitment in the tumor microenvironment. At the same time, IFN-γ can induce IDO1 activity in tumor cells, which activates the JNK pathway, leading to p53 downregulation and tumor growth. Finally, the concomitant stimulation of tumor-specific CD8+ T cells by ICI therapy and T-cell-receptor (TCR) activation results in a hyperactivated immune environment in which IFN-γ triggers the activation-induced cell death (AICD) mechanism and T-cell Fas-mediated apoptosis.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13020309
References
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11.
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030.
- Ahamadi, M.; Freshwater, T.; Prohn, M.; Li, C.; de Alwis, D.; de Greef, R.; Elassaiss-Schaap, J.; Kondic, A.; Stone, J. Model-Based Characterization of the Pharmacokinetics of Pembrolizumab: A Humanized Anti-PD-1 Monoclonal Antibody in Advanced Solid Tumors. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 49–57.
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738.
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330.
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550.
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396.
- Kamada, T.; Togashi, Y.; Tay, C.; Ha, D.; Sasaki, A.; Nakamura, Y.; Sato, E.; Fukuoka, S.; Tada, Y.; Tanaka, A.; et al. PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 9999–10008.
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501.
- Huang, R.-Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. OncoImmunology 2017, 6, e1249561.
- Zuazo-Ibarra, M.; Arasanz, H.; Fernández-Hinojal, G.; María, G.-C.; Hernández-Marín, B.; Martínez-Aguillo, M.; Jose Lecumberri, M.; Fernández, A.; Teijeira, L.; Vera, R.; et al. Highly differentiated CD4 T cells Unequivocally Identify Primary Resistance and Risk of Hyperprogression to PD-L1/PD-1 Immune Checkpoint Blockade in Lung Cancer. bioRxiv 2018, 320176.
- Kato, S.; Goodman, A.; Walavalkar, V.; Barkauskas, D.A.; Sharabi, A.; Kurzrock, R. Hyperprogressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin. Cancer Res. 2017, 23, 4242–4250.
- Stein, R.G.; Ebert, S.; Schlahsa, L.; Scholz, C.J.; Braun, M.; Hauck, P.; Horn, E.; Monoranu, C.-M.; Thiemann, V.J.; Wustrow, M.P.; et al. Cognate Nonlytic Interactions between CD8 + T Cells and Breast Cancer Cells Induce Cancer Stem Cell–like Properties. Cancer Res. 2019, 79, 1507–1519.
- Du, S.; McCall, N.; Park, K.; Guan, Q.; Fontina, P.; Ertel, A.; Zhan, T.; Dicker, A.P.; Lu, B. Blockade of Tumor-Expressed PD-1 promotes lung cancer growth. OncoImmunology 2018, 7, e1408747.
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 2008, 111, 3635–3643.
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241.
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974.
- Gato-Cañas, M.; Zuazo, M.; Arasanz, H.; Ibañez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829.
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452.
- Lamichhane, P.; Karyampudi, L.; Shreeder, B.; Krempski, J.; Bahr, D.; Daum, J.; Kalli, K.R.; Goode, E.L.; Block, M.S.; Cannon, M.J.; et al. IL10 Release upon PD-1 Blockade Sustains Immunosuppression in Ovarian Cancer. Cancer Res. 2017, 77, 6667–6678.
- Sun, Z.; Fourcade, J.; Pagliano, O.; Chauvin, J.-M.; Sander, C.; Kirkwood, J.M.; Zarour, H.M. IL10 and PD-1 Cooperate to Limit the Activity of Tumor-Specific CD8+ T Cells. Cancer Res. 2015, 75, 1635–1644.
- Scholz, A.; Lang, V.; Henschler, R.; Czabanka, M.; Vajkoczy, P.; Chavakis, E.; Drynski, J.; Harter, P.N.; Mittelbronn, M.; Dumont, D.J.; et al. Angiopoietin-2 promotes myeloid cell infiltration in a β2-integrin–dependent manner. Blood 2011, 118, 5050–5059.
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Connelly, C.; Connolly, E.M.; Li, J.; Manos, M.P.; Lawrence, D.; McDermott, D.; Severgnini, M.; et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol. Res. 2017, 5, 17–28.
- Lo Russo, G.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin. Cancer Res. 2019, 25, 989–999.
- Dulos, J.; Carven, G.J.; van Boxtel, S.J.; Evers, S.; Driessen-Engels, L.J.A.; Hobo, W.; Gorecka, M.A.; de Haan, A.F.J.; Mulders, P.; Punt, C.J.A.; et al. PD-1 Blockade Augments Th1 and Th17 and Suppresses Th2 Responses in Peripheral Blood From Patients With Prostate and Advanced Melanoma Cancer. J. Immunother. 2012, 35, 169–178.
- Chin, Y.E.; Kitagawa, M.; Kuida, K.; Flavell, R.A.; Fu, X.Y. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol. Cell. Biol. 1997, 17, 5328–5337.
- Xu, X.; Fu, X.-Y.; Plate, J.; Chong, A.S.-F. IFN-y Induces Cell Growth Inhibition by Fas-mediated Apoptosis: Requirement of STATI Protein for Up-Regulation of Fas and FasL Expression. Cancer Res. 1998, 58, 2832–2837.
- Hobeika, A.C.; Etienne, W.; Torres, B.A.; Johnson, H.M.; Subramaniam, P.S. IFN-gamma Induction of p21WAF1 Is Required for Cell Cycle Inhibition and Suppression of Apoptosis. J. Interferon Cytokine Res. 1999, 19, 1351–1361.
- Fulda, S.; Debatin, K.-M. IFNγ sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene 2002, 21, 2295–2308.
- Kammertoens, T.; Friese, C.; Arina, A.; Idel, C.; Briesemeister, D.; Rothe, M.; Ivanov, A.; Szymborska, A.; Patone, G.; Kunz, S.; et al. Tumour ischaemia by interferon-γ resembles physiological blood vessel regression. Nature 2017, 545, 98–102.
- Lollini, P.-L.; de Giovanni, C.; del Re, B.; Nicoletti, G.; Prodi, G.; Nanni, P. Interferon-mediated enhancement of metastasis. Are MHC antigens involved? Clin. Exp. Metastasis 1987, 5, 277–287.
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847.
- Zhang, Y.; Apilado, R.; Coleman, J.; Ben-Sasson, S.; Tsang, S.; Hu-Li, J.; Paul, W.E.; Huang, H. Interferon γ Stabilizes the T Helper Cell Type 1 Phenotype. J. Exp. Med. 2001, 194, 165–172.
- Curtsinger, J.M.; Agarwal, P.; Lins, D.C.; Mescher, M.F. Autocrine IFN-γ Promotes Naive CD8 T Cell Differentiation and Synergizes with IFN-α To Stimulate Strong Function. J. Immunol. 2012, 189, 659–668.
- Overacre-Delgoffe, A.E.; Chikina, M.; Dadey, R.E.; Yano, H.; Brunazzi, E.A.; Shayan, G.; Horne, W.; Moskovitz, J.M.; Kolls, J.K.; Sander, C.; et al. Interferon-γ Drives T reg Fragility to Promote Anti-tumor Immunity. Cell 2017, 169, 1130–1141.
- Mojic, M.; Takeda, K.; Hayakawa, Y. The Dark Side of IFN-γ: Its Role in Promoting Cancer Immunoevasion. Int. J. Mol. Sci. 2017, 19, 89.
- Lollini, P.-L.; Nanni, P.; de Giovanni, C.; Nicoletti, G.; Landuzzi, L. Re: Randomized Trial of Adjuvant Human Interferon Gamma Versus Observation in High-Risk Cutaneous Melanoma: A Southwest Oncology Group Study. JNCI J. Natl. Cancer Inst. 1996, 88, 926–927.
- Lollini, P.-L.; Bosco, M.C.; Cavallo, F.; de Giovanni, C.; Giovarelli, M.; Landuzzi, L.; Musiani, P.; Modesti, A.; Nicoletti, G.; Palmieri, G.; et al. Inhibition of tumor growth and enhancement of metastasis after transfection of the γ-interferon gene. Int. J. Cancer 1993, 55.
- Ribas, A.; Butler, M.; Lutzky, J.; Lawrence, D.P.; Robert, C.; Miller, W.; Linette, G.P.; Ascierto, P.A.; Kuzel, T.; Algazi, A.P.; et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J. Clin. Oncol. 2015, 33, 3003.
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201.
- Lamberti, G.; Sisi, M.; Andrini, E.; Palladini, A.; Giunchi, F.; Lollini, P.-L.; Ardizzoni, A.; Gelsomino, F. The Mechanisms of PD-L1 Regulation in Non-Small-Cell Lung Cancer (NSCLC): Which Are the Involved Players? Cancers 2020, 12, 3129.
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829.
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418.
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39.
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-γ Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515.
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12.
- Lu, Y.; Gu, X.; Chen, L.; Yao, Z.; Song, J.; Niu, X.; Xiang, R.; Cheng, T.; Qin, Z.; Deng, W.; et al. Interferon-γ produced by tumor-infiltrating NK cells and CD4+ T cells downregulates TNFSF15 expression in vascular endothelial cells. Angiogenesis 2014, 17, 529–540.
- Jauch, D.; Martin, M.; Schiechl, G.; Kesselring, R.; Schlitt, H.J.; Geissler, E.K.; Fichtner-Feigl, S. Interleukin 21 controls tumour growth and tumour immunosurveillance in colitis-associated tumorigenesis in mice. Gut 2011, 60, 1678–1686.
- Carbotti, G.; Barisione, G.; Airoldi, I.; Mezzanzanica, D.; Bagnoli, M.; Ferrero, S.; Petretto, A.; Fabbi, M.; Ferrini, S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 2015, 6, 43267–43280.
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A.A. Interleukin-35 Limits Anti-Tumor Immunity. Immunity 2016, 44, 316–329.
- Zhao, Z.; Chen, X.; Hao, S.; Jia, R.; Wang, N.; Chen, S.; Li, M.; Wang, C.; Mao, H. Increased interleukin-35 expression in tumor-infiltrating lymphocytes correlates with poor prognosis in patients with breast cancer. Cytokine 2017, 89, 76–81.
- Zou, J.-M.; Qin, J.; Li, Y.-C.; Wang, Y.; Li, D.; Shu, Y.; Luo, C.; Wang, S.-S.; Chi, G.; Guo, F.; et al. IL-35 induces N2 phenotype of neutrophils to promote tumor growth. Oncotarget 2017, 8, 33501–33514.
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035.
- Huang, B.; Pan, P.-Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.-H. Gr-1+ CD115+ Immature Myeloid Suppressor Cells Mediate the Development of Tumor-Induced T Regulatory Cells and T-Cell Anergy in Tumor-Bearing Host. Cancer Res. 2006, 66, 1123–1131.
- Nishibori, T.; Tanabe, Y.; Su, L.; David, M. Impaired Development of CD4+ CD25+ Regulatory T Cells in the Absence of STAT1. J. Exp. Med. 2004, 199, 25–34.
- Wang, Z.; Hong, J.; Sun, W.; Xu, G.; Li, N.; Chen, X.; Liu, A.; Xu, L.; Sun, B.; Zhang, J.Z. Role of IFN-g in induction of Foxp3 and conversion of CD4+CD25- T cells to CD4+ Tregs. J. Clin. Investig. 2006, 116, 2434–2441.
- Sawitzki, B.; Kingsley, C.I.; Oliveira, V.; Karim, M.; Herber, M.; Wood, K.J. IFN-γ production by alloantigen-reactive regulatory T cells is important for their regulatory function in vivo. J. Exp. Med. 2005, 201, 1925–1935.
- Wei, B.; Baker, S.; Wieckiewicz, J.; Wood, K.J. IFN-γ Triggered STAT1-PKB/AKT Signalling Pathway Influences the Function of Alloantigen Reactive Regulatory T Cells. Am. J. Transplant. 2010, 10, 69–80.
- Feng, T.; Cao, A.T.; Weaver, C.T.; Elson, C.O.; Cong, Y. Interleukin-12 Converts Foxp3+ Regulatory T Cells to Interferon–γ-Producing Foxp3+ T Cells That Inhibit Colitis. Gastroenterology 2011, 140, 2031–2043.
- Koenecke, C.; Lee, C.-W.; Thamm, K.; Föhse, L.; Schafferus, M.; Mittrücker, H.-W.; Floess, S.; Huehn, J.; Ganser, A.; Förster, R.; et al. IFN-γ Production by Allogeneic Foxp3 + Regulatory T Cells Is Essential for Preventing Experimental Graft-versus-Host Disease. J. Immunol. 2012, 189, 2890–2896.
- Greifenberg, V.; Ribechini, E.; Rößner, S.; Lutz, M.B. Myeloid-derived suppressor cell activation by combined LPS and IFN-γ treatment impairs DC development. Eur. J. Immunol. 2009, 39, 2865–2876.
- Shime, H.; Maruyama, A.; Yoshida, S.; Takeda, Y.; Matsumoto, M.; Seya, T. Toll-like receptor 2 ligand and interferon-γ suppress anti-tumor T cell responses by enhancing the immunosuppressive activity of monocytic myeloid-derived suppressor cells. OncoImmunology 2018, 7, e1373231.
- Weide, B.; Martens, A.; Zelba, H.; Stutz, C.; Derhovanessian, E.; di Giacomo, A.M.; Maio, M.; Sucker, A.; Schilling, B.; Schadendorf, D.; et al. Myeloid-Derived Suppressor Cells Predict Survival of Patients with Advanced Melanoma: Comparison with Regulatory T Cells and NY-ESO-1- or Melan-A-Specific T Cells. Clin. Cancer Res. 2014, 20, 1601–1609.
- Sade-Feldman, M.; Kanterman, J.; Klieger, Y.; Ish-Shalom, E.; Olga, M.; Saragovi, A.; Shtainberg, H.; Lotem, M.; Baniyash, M. Clinical Significance of Circulating CD33+CD11b+HLA-DR- Myeloid Cells in Patients with Stage IV Melanoma Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 5661–5672.
- Martens, A.; Wistuba-Hamprecht, K.; Foppen, M.G.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918.
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257.
- Faure, M.; Rochigneux, P.; Olive, D.; Taix, S.; Brenot-Rossi, I.; Gilabert, M. Hyperprogressive Disease in Anorectal Melanoma Treated by PD-1 Inhibitors. Front. Immunol. 2018, 9, 797.
- Xiong, D.; Wang, Y.; Singavi, A.K.; Mackinnon, A.C.; George, B.; You, M. Immunogenomic Landscape Contributes to Hyperprogressive Disease after Anti-PD-1 Immunotherapy for Cancer. iScience 2018, 9, 258–277.
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.S.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti–PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586.
- Ito, S.; Hara, Y.; Kubota, T. CARD8 is a negative regulator for NLRP3 inflammasome, but mutant NLRP3 in cryopyrin-associated periodic syndromes escapes the restriction. Arthritis Res. Ther. 2014, 16, R52.
- Burger, D.; Fickentscher, C.; de Moerloose, P.; Brandt, K.J. F-actin dampens NLRP3 inflammasome activity via Flightless-I and LRRFIP2. Sci. Rep. 2016, 6, 29834.
- Clipman, S.J.; Henderson-Frost, J.; Fu, K.Y.; Bern, C.; Flores, J.; Gilman, R.H. Genetic association study of NLRP1, CARD, and CASP1 inflammasome genes with chronic Chagas cardiomyopathy among Trypanosoma cruzi seropositive patients in Bolivia. PLoS ONE 2018, 13, e0192378.
- Hennig, P.; Garstkiewicz, M.; Grossi, S.; di Filippo, M.; French, L.; Beer, H.D. The Crosstalk between Nrf2 and Inflammasomes. Int. J. Mol. Sci. 2018, 19, 562.
- Yue, S.; Li, C.; Ke, M.; Lu, L.; Busuttil, R.; Ying, Q.; Kupiec-Weglinski, J.; Ke, B. Notch Signal Regulates Inflammasome NLRP3 Activation in Liver Ischemia and Reperfusion Injury. J. Hepatol. 2016, 64, S508–S509.
- Jiang, L.; Ke, M.; Yue, S.; Xiao, W.; Yan, Y.; Deng, X.; Ying, Q.L.; Li, J.; Ke, B. Blockade of Notch signaling promotes acetaminophen-induced liver injury. Immunol. Res. 2017, 65, 739–749.
- Jin, Y.; Li, C.; Xu, D.; Zhu, J.; Wei, S.; Zhong, A.; Sheng, M.; Duarte, S.; Coito, A.J.; Busuttil, R.W.; et al. Jagged1-mediated myeloid Notch1 signaling activates HSF1/Snail and controls NLRP3 inflammasome activation in liver inflammatory injury. Cell. Mol. Immunol. 2019, 17.
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310.
- Holtzhausen, A.; Zhao, F.; Evans, K.S.; Tsutsui, M.; Orabona, C.; Tyler, D.S.; Hanks, B.A. Melanoma-Derived Wnt5a Promotes Local Dendritic-Cell Expression of IDO and Immunotolerance: Opportunities for Pharmacologic Enhancement of Immunotherapy. Cancer Immunol. Res. 2015, 3, 1082–1095.
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-β-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160.
- Badawy, A.A.B. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10, 117864691769193.
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The Role of Indoleamine-2,3-Dioxygenase in Cancer Development, Diagnostics, and Therapy. Front. Immunol. 2018, 9, 151.
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274.
- Katz, J.B.; Muller, A.J.; Prendergast, G.C. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol. Rev. 2008, 222, 206–221.
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207.
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642.
- Metz, R.; Rust, S.; DuHadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. OncoImmunology 2012, 1, 1460–1468.
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761.
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198.
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33.
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100.
- Taube, J.M.; Young, G.D.; McMiller, T.L.; Chen, S.; Salas, J.T.; Pritchard, T.S.; Xu, H.; Meeker, A.K.; Fan, J.; Cheadle, C.; et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clin. Cancer Res. 2015, 21, 3969–3976.
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56.
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells. Sci. Transl. Med. 2013, 5.
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25− into CD25+ T regulatory cells. Blood 2007, 109, 2871–2877.
- Yu, G.; Dai, H.; Chen, J.; Duan, L.; Gong, M.; Liu, L.; Xiong, P.; Wang, C.Y.; Fang, M.; Gong, F. Gene delivery of indoleamine 2,3-dioxygenase prolongs cardiac allograft survival by shaping the types of T-cell responses. J. Gene Med. 2008, 10, 754–761.
- Witkiewicz, A.; Williams, T.K.; Cozzitorto, J.; Durkan, B.; Showalter, S.L.; Yeo, C.J.; Brody, J.R. Expression of Indoleamine 2,3-Dioxygenase in Metastatic Pancreatic Ductal Adenocarcinoma Recruits Regulatory T Cells to Avoid Immune Detection. J. Am. Coll. Surg. 2008, 206, 849–854.
- Mansfield, A.S.; Heikkila, P.S.; Vaara, A.T.; von Smitten, K.A.J.; Vakkila, J.M.; Leidenius, M.H.K. Simultaneous Foxp3 and IDO expression is associated with sentinel lymph node metastases in breast cancer. BMC Cancer 2009, 9, 231.
- Baban, B.; Chandler, P.R.; Sharma, M.D.; Pihkala, J.; Koni, P.A.; Munn, D.H.; Mellor, A.L. IDO Activates Regulatory T Cells and Blocks Their Conversion into Th17-Like T Cells. J. Immunol. 2009, 183, 2475–2483.
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.C.; et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928.
- Botticelli, A.; Cerbelli, B.; Lionetto, L.; Zizzari, I.; Salati, M.; Pisano, A.; Federica, M.; Simmaco, M.; Nuti, M.; Marchetti, P. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J. Transl. Med. 2018, 16, 219.
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402.
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3.
- Perez, R.P.; Riese, M.J.; Lewis, K.D.; Saleh, M.N.; Daud, A.; Berlin, J.; Lee, J.J.; Mukhopadhyay, S.; Zhou, L.; Serbest, G.; et al. Epacadostat plus nivolumab in patients with advanced solid tumors: Preliminary phase I/II results of ECHO-204. J. Clin. Oncol. 2017, 35, 3003.
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230.
- Gibney, G.T.; Hamid, O.; Lutzky, J.; Olszanski, A.J.; Mitchell, T.C.; Gajewski, T.F.; Chmielowski, B.; Hanks, B.A.; Zhao, Y.; Newton, R.C.; et al. Phase 1/2 study of epacadostat in combination with ipilimumab in patients with unresectable or metastatic melanoma. J. Immunother. Cancer 2019, 7, 80.
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat (E) plus pembrolizumab (P) versus pembrolizumab alone in patients (pts) with unresectable or metastatic melanoma: Results of the phase 3 ECHO-301/KEYNOTE-252 study. J. Clin. Oncol. 2018, 36, 108.
- Van den Eynde, B.J.; van Baren, N.; Baurain, J.F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256.
- Tang, D.; Yue, L.; Yao, R.; Zhou, L.; Yang, Y.; Lu, L.; Gao, W. P53 prevent tumor invasion and metastasis by down-regulating IDO in lung cancer. Oncotarget 2017, 8, 54548–54557.
- Mei, J.; Li, M.-Q.; Ding, D.; Li, D.-J.; Jin, L.-P.; Hu, W.-G.; Zhu, X.-Y. Indoleamine 2,3-dioxygenase-1 (IDO1) enhances survival and invasiveness of endometrial stromal cells via the activation of JNK signaling pathway. Int. J. Clin. Exp. Pathol. 2013, 6, 431–444.
- Pallotta, M.T.; Orabona, C.; Volpi, C.; Vacca, C.; Belladonna, M.L.; Bianchi, R.; Servillo, G.; Brunacci, C.; Calvitti, M.; Bicciato, S.; et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat. Immunol. 2011, 12, 870–878.
- Hammaker, D.R.; Boyle, D.L.; Inoue, T.; Firestein, G.S. Regulation of the JNK pathway by TGF-beta activated kinase 1 in rheumatoid arthritis synoviocytes. Arthritis Res. Ther. 2007, 9, R57.
- Wu, S.; Kasisomayajula, K.; Peng, J.; Bancalari, E. Inhibition of JNK Enhances TGF-β1-Activated Smad2 Signaling in Mouse Embryonic Lung. Pediatric Res. 2009, 65, 381–386.
- Hennequart, M.; Pilotte, L.; Cane, S.; Hoffmann, D.; Stroobant, V.; Plaen, E.; van den Eynde, B.J. Constitutive IDO1 Expression in Human Tumors Is Driven by Cyclooxygenase-2 and Mediates Intrinsic Immune Resistance. Cancer Immunol. Res. 2017, 5, 695–709.
- Xu, Z.; Chen, L.; Zheng, L.; Yang, Q.; Chen, M.; Wang, J.; Zhu, G.; Chen, Z.; Sun, J. Hyperprogressive Disease In Cervical Small Cell Carcinoma Treated By Immune Checkpoint Inhibitor. Oncotargets Ther. 2019, 12, 8873–8877.
- Pai, C.-C.S.; Huang, J.T.; Lu, X.; Simons, D.M.; Park, C.; Chang, A.; Tamaki, W.; Liu, E.; Roybal, K.T.; Seagal, J.; et al. Clonal Deletion of Tumor-Specific T Cells by Interferon-γ Confers Therapeutic Resistance to Combination Immune Checkpoint Blockade. Immunity 2019, 50, 477–492.
- Refaeli, Y.; van Parijs, L.; Alexander, S.I.; Abbas, A.K. Interferon γ is Required for Activation-induced Death of T Lymphocytes. J. Exp. Med. 2002, 196, 999–1005.
- Ju, S.-T.; Panka, D.J.; Cui, H.; Ettinger, R.; EI-Khatib, M.; Sherr, D.H.; Stanger, B.Z.; Marshak-Rothstein, A. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 1995, 373, 444–448.