NF-κB is a family of five eukaryotic transcription factors, p50, p52, p65 (RelA), RelB and c-Rel, which form 15 different homodimers and heterodimers.
- nuclear factor kappa B
- preeclampsia
- pregnancy
- inflammation
1. Nuclear Factor Kappa B
NF-κB was first identified as a transcription factor regulating expression of immunoglobulin light chain kappa in lymphocytes B; however, soon it was discovered that it controls transcription of many other genes, and after over 30 years of research, over 150 of them can be named [1]. NF-κB is a family of five eukaryotic transcription factors, p50, p52, p65 (RelA), RelB and c-Rel, encoded by NFKB1, NFKB2, RELA, RELB, and REL genes, respectively [1][2][3]. These proteins do not act solitarily, but form 15 different homodimers and heterodimers, which are translocated into the nucleus and bind to a specific set of related 9–11 bp DNA sites, collectively called kB sites [1]. All proteins have the N-terminal Rel homology domain, which is responsible for both dimerization and binding to κB sites [2]. However, only RelA, RelB, and c-Rel have the transcription activation domain (TAD), which is essential for positive regulation of gene expression [2]. Therefore, p50 and p52 homodimers, as they lack TADs, may downregulate expression of genes with κB sites until they are replaced by NF-κB dimers with TADs. This is only a small part of complexity of gene expression regulation by NF-κB, which allows it to interfere with more than 150 of them. Interestingly, the N-terminal part of Rel homology domain slightly differs between members of NF-κB family, which results in preferential formation of heterodimers rather than homodimers [4]. Furthermore, Rel homology domains make base-specific and non-specific contacts with DNA, and aforementioned variations result in different affinity to particular genes, depending on number of their DNA base pairs (9–11) in κB sites [4].
In resting cells, NF-κB is stored in the cytoplasm in its inactive form as dimers associated with inhibitors of nuclear factor kappa B (IκB) or precursor proteins p100 and p105, which prevents NF-κB from entering into the nucleus [1][2][3]. Proteins p100 and p105 are precursor proteins of p52 and p50, respectively, and if cleavaged to their active form they facilitate transportation into the nucleus and DNA binding. There are three typical types of IκB proteins, IκBα, IκBβ, and IκBε, and three types of inducibly expressed, atypical IκB, Bcl-3, and IκBζ [1][2][3]. Every NF-κB has the nuclear localization sequence (NLS), which facilitates its transportation into the nucleus; however, it is covered by IκBs [2][3]. Therefore, activation of NF-κB requires its release from IκB, and predominantly this process is mediated by a complex of IκB kinases (IKK), IKKα, IKKβ, and IKKγ, which leads to phosphorylation, ubiquitination, and degradation of IκB by proteasome [3].
There are three known NF-κB activation pathways: canonical, non-canonical, and atypical [3][4][5][6].
The canonical pathway leads to activation of NF-κB through degradation of IκBs by IKK complex [3][4][5]. The activators of this pathway are interleukin 1 (IL-1) and a vast number of different damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), such as high-mobility group box 1 (HMGB1), fibrinogen, extracellular DNA, ATP and histones, glycans, flagellin, lipoteichoic acid, etc. IL-1 is a ligand for IL-1 receptors (IL-1Rs) and activated IL-1R recruits the interleukin-1-receptor-associated kinase (IRAK-1) through the adapter molecule—myeloid differentiation primary response 88 (MYD88) [7][8][9]. Recruited IRAK-1 is highly phosphorylated and dissociates form the receptor into the cytoplasm and associates with IKKα–IKKβ–IKKγ complexes, where it activates IKKγ, also termed the NF-κB essential modulator (NEMO). Activated NEMO oligomerize and initiate autophosphorylation of IKKs, hereby activating the complex [2][7][8]. Active IKK complex phosphorylates and leads to degradation of IκBs, therefore, leads to release and activation of NF-κB. Interestingly, active IRAK-1 is also translocated into the nucleus, where it associates with promoters of NF-κB regulated genes increasing their expression, inter alia, the expression of IκBα [7]. Thereby, IRAK-1 mediates not only the activation of NF-κB, but also regulates its inhibition. Other receptors involved in the NF-κB activation are toll-like receptors (TLRs), whose ligands are DAMPs and PAMPs, and the whole process is identical, as in the case of IL-1Rs [7].
The non-canonical pathway can be activated by the tumor necrosis factor (TNF), B-cell activating factor (BAFF), CD40 ligands, or virus RNA [2][5][7]. Activated BAFF receptor (BAFFR), CD40, lymphotoxin β receptor (LTβR), or receptor activator for NF-κB (RANKL) associates with TNF receptor associated factors (TRAFs) complexes, which in turn release the NF-κB-inducing kinase (NIK) [10]. NIK activates IKKα, which forms homodimers and promotes IKKα association with p100 in p100/RelB heterodimers. Then NIK phosphorylates p100, which leads to its ubiquitination and processing in proteasome into p52 [10]. Active p52/RelB heterodimer is translocated into the nucleus where it regulates gene expression.
The atypical pathway is not one mechanism of NF-κB activation, but rather a term used for any of them that is different from canonical and non-canonical pathways and which is often not fully comprehended [5][6]. However, most often it involves casein kinase 2 (CK2), which is activated by reactive oxygen species, hypoxia, UV, or DNA damage phosphorylase IκBs, leading to its degradation in proteasome and release of NF-κB [5][6]. However, the exact mechanism of this process is unknown.
The variety of NF-κB activating factors, activation pathways, and regulated genes contribute to the complexity of NF-κB signaling and its involvement in different processes. One of them is a remodeling of uterine spiral arteries during pregnancy, and the crucial role of NF-κB in that process is described in the following section.
2. NF-κB as a Mediator of Spiral Arteries Remodeling
2.1. General Information
Considering that NF-κB is an essential mediator for many processes, it is no surprise that it appears to be a key regulator during pregnancy, taking part in its development, maintenance, and termination. Within the first days after the conception, NF-κB regulates time of the implantation through induction of leukemia inhibitory factor (LIF) production and inflammation, which results in removal of mucins covering adhesion molecules present on the surface of uterine epithelium [11][12]. Furthermore, inflammation is associated with increased expression of L-selectin on the surface of uterine epithelial cells, facilitating blastocyst adhesion and implantation [11]. Inflammation was reported to be characteristic for the implantation period, and the first trimester of pregnancy in general is associated with increased production of many signaling molecules, such as TNFα, chemokine (C-X-C motif)-ligand 1 (CXCL1), IL-1, IL-2, IL-8, and IL-15, whose expression is controlled by NF-κB [1][13][14][15][16]. Mor et al. indicated that embryo implantation presents “an open wound” phenotype, which further increases NF-κB activation [15]. Inflammatory state in uterus stimulates local white blood cells such as dendritic cells and uterine natural killer cells (uNK) and also leads to infiltration of the implantation site by new monocytes, lymphocytes, and neutrophils [17][18]. Interestingly, inflammation, associated activation of leukocytes, and initial changes in spiral arteries have been reported to occur before the implantation; therefore, embryo antigens are not initiators of inflammation in the uterus, which is crucial for embryo adhesion and implantation [19][20]. Researchers hypothesized that this is a result of hormonal changes during the menstrual cycle; however, an important trigger for uterine inflammation appear to be insemination [19][20].
2.2. Insemination as an Inflammation Trigger
Semen is not simply a carrier of spermatozoa, but it has various functions like increase of pH, facilitating the movement of spermatozoa, etc. [19][20][21]. For decades, it has been the subject of research, and knowledge of its role in fertilization and embryo development has grown steadily. First trials with in vitro fertilization in rodent models showed that embryo transfers to females not exposed to male fluids were associated with greater rates of implantation failure, miscarriage, foetal growth restriction, and abnormalities [22][23]. Similar findings have been made regarding human embryos [24][25]. Therefore, investigation of seminal fluid components followed.
Many studies reported that semen induces inflammatory response in the uterus through specific mediators and presentation of paternal antigens [19][20][21][26]. The most important seminal cytokine is transforming growth factor β (TGFβ), which stimulates uterine epithelial cells to production of proinflammatory cytokines, such as granulocyte-macrophage colony stimulating factor (GM-CSF), colony stimulating factor 1/macrophage colony stimulating factor (CFS1)/(M-CSF), IL-1α, IL-6, IL-8, LIF, RANTES, and macrophage inflammatory protein 1α (MIP-1α) [19][20][21]. This process is most likely mediated through activation of the TGFβ receptor I and II (TGFβRI/TGFβRII) complex, which in turn leads to the activation of NF-κB; however, this process is not fully comprehended [27]. Torrealba et al. examined the expression of TGFβ, TGFβRI, TGFβRII, NF-κB, PI3K, AKT, and mTOR in specimens collected from 106 patients with prostatic cancer [28]. Analysis revealed increased expression of TGFβ, PI3K/AKT/mTOR pathway compounds, IKK, and NF-κB [28]. Relating these findings to previous studies, researchers assumed that TGFβ through TGFβRs activates the PI3K/AKT/mTOR pathway, which in turn activates IKK complexes, thus NF-κB [28][29]. This process is possibly responsible for induction of proinflammatory cytokine production by uterine epithelium, as GM-CSF, M-CSF, IL-1α, IL-6, IL-8, LIF, RANTES, and MIP-1α gene expression is regulated by NF-κB [1][12]. Robertson et al. indicated that these mediators induce inflammation in uterus and activate uterine dendritic cells (DCs) and macrophages, and the latter leads to further stimulation of DC and inflammation spreading through TNFα release [20]. Moreover, aforementioned cytokines lead to substantial recruitment and activation of circulating monocytes and granulocytes [20]. Activated DCs have an important role, as they act as antigen presenting cells (APCs) presenting paternal MHC antigens to lymphocytes T, which results in their hypo-responsiveness for paternal antigens and fetal cell immunotolerance [20][26]. As was mentioned in the previous section, inflammation leads to removal of mucins covering epithelium and presentation of adhesion molecules e.g., L-selectin [11]. Moreover, infiltrating macrophages and neutrophils produce various metalloproteinases (MMPs), inter alia MMP-2 and MMP-9, which disintegrate the extracellular matrix (ECM) in order to facilitate implantation and trophoblast invasion [30][31]. Interestingly, it has been reported that MMP-9 production in breast cancer cells in regulated by PI3K/AKT/NF-κB pathway; therefore, it can be assumed that MMP-9 release by macrophages and neutrophils is also controlled by NF-κB [32]. These findings indicate that NF-κB is a key mediator of the implantation process. Alongside macrophages, uterine natural killer cells (uNKs) are the most prominent type of leukocytes in the placental bed [33]. They are activated by HLA class I antigens, HLA-C, HLA-E, and HLA-G presented by trophoblast and regulate spiral arteries remodeling and placentation [33][34][35][36][37]. The trophoblast invasion and uNK activation starts a sophisticated crosstalk between macrophages, uNKs, and trophoblasts, on which the future of pregnancy depends.
2.3. Trophoblast/uNK/Macrophage Crosstalk
Uterine NKs are different from circulating NKs and are called CD56bright, as they show increased expression of CD56 [38]. This type of NKs was reported to be less cytotoxic and to produce more cytokines than other NK types; thus, it was assumed that CD56bright have a regulatory function in inflammation [33][36][38]. In a mouse model, Chkraborty et al. have proven that the presence of uNKs is crucial for spiral arteries remodeling [39]. Uterine NK cell depletion was achieved by treatment with anti-asialo GM1 antibodies on gestation day 4.5 or 4.5 and 9.5. Placentation sites were collected and examined on day 9.5 or 13.5 [39]. The disruption of the vascular smooth muscle layer of spiral arteries was observed only in samples with uNKs and was abolished in samples lacking uNKs [20]. Furthermore, trophoblast invasion into the uterine wall was much deeper and chaotic in uNK deficient uteri [39]. Interestingly, uNKs modified trophoblast phenotype led researchers to conclusions that uNKs control disorganization of spiral arteries’ smooth muscle layers, modulate trophoblast phenotype, and restrain its invasion [39]. Zhang and Tian proposed a mechanism of uNK function in which killer-cell immunoglobulin-like receptors expressed by uNK recognize MHCs presented by trophoblasts [36]. This leads to the activation of uNKs and production of various factors, such as IL-8 and CXCL-10, which in turn affect trophoblasts through CXCR1 and CXCR3 receptors, and this is most likely the mechanism of trophoblast phenotype and function modification by uNKs [22]. In response to that, stimulation trophoblasts produce TGF-β, chorionic gonadotropin (HCG), and VEGF [36]. The other factors produced by activated uNKs are M-CSF, GM-CSF, TNF-α, IFN-γ, TGF-β1, VEGF, PlGF, LIF, angiopoietin 1 (Ang-1), Ang-2, MMP-2, and MMP-9 [33][36][40][41]. These data indicate that uNKs and trophoblasts enhance the embryo related inflammatory response in uterus, leading to further activation of uNKs and trophoblasts. Liu et al. reported that TGF-β1 has a direct impact on vascular smooth muscle cells (VSMCs) through maternally expressed gene 3 (MEG-3) and induces VSMCs apoptosis and migration and suppresses their proliferation [37]. These effects of TGF-β1 were abolished in MEG-3 silenced VMSC cultures [37].
Other cells important for spiral arteries remodeling and placentation are macrophages. Some authors indicated that in the remodeling process, macrophages M2 are the majority of macrophages infiltrating the spiral arteries site; however, it appears that these macrophages are hard to define, as there can be many intermediate types between proinflammatory type M1 and anti-inflammatory M2 [42]. It is most likely a result of a great variety of aforementioned signaling factors associated with implantation related inflammation. For example, TGF-β, IL-6, and VEGF are simulating M2 polarization, while TNF-α and IFN-γ are stimulating M1 polarization, and combination of these factors may result in formation of atypical macrophages [43][44]. Nevertheless, it appears that there are more M2 macrophages in uterus during spiral arteries remodeling [42][45]. Macrophages activated by cytokines and other factors produced by endometrium epithelial cells, uNKs, and trophoblasts infiltrate decidua around spiral arteries and secrete a wide range of cytokines, IL-1β, IL-4, IL-6, IL-8, IL-10, IL-13, and TNF-α, which lead to further extension of inflammatory response and stimulation of uNKs [42][45]. However, the most important function of macrophages is the secretion of MMP-1, MMP-2, MMP-7, MMP-9, and MMP-10, disorganization of the extracellular matrix, and phagocytosis of apoptotic VSMCs and trophoblasts [33][42][45][46]. Although it was reported that macrophages have no impact on VSMCs migration and apoptosis, the disorganization of the extracellular matrix facilitates that process, and phagocytosis of post-apoptotic cell rests decreases DAMP release, which alongside with secretion of IL-10 restrains excessive inflammatory response, which could dysregulate the remodeling process [33][42][45][46]. As it was mentioned before, uNKs secrete TNF-α and IFN-γ, which are essential to stimulate uterine macrophages to produce CXCL10 [47]. Another CXCL10 production stimulator is hypoxia, which frequently occurs in the uterus during the placentation [48][49]. In such environment macrophages, infiltrating spiral arteries produce CXCL10, which is a potent chemoattractive molecule for trophoblasts [50]. Therefore, during the placentation, macrophages not only facilitate but also stimulate spiral arteries invasion by trophoblasts. Finally, monocytes stimulated by GM-CFS differentiate into dendritic cells (DCs), which through secretion of soluble FMS-like protein 1 (sFlt-1) and TGF-β1 play a pivotal role in placentation related angiogenesis [43][51]. As a result of the described processes, VSMCs detach from the spiral arteries’ wall, undergo apoptosis, and become phagocytosed by macrophages, while extravillous trophoblasts take their place in the spiral artery wall [18]. This process, strictly controlled by trophoblast/uNK/macrophage crosstalk, results in the formation of wide, cone-like endings of spiral arteries, in which maternal blood flows around foetal capillaries and exchanges oxygen, carbon dioxide, nutrients, etc. [18]. This complex and fragile process is a basis of pregnancy; its disturbance can have dread consequences and is regulated by the trophoblast/uNK/macrophage subtle crosstalk as it is summarized in Figure 1.
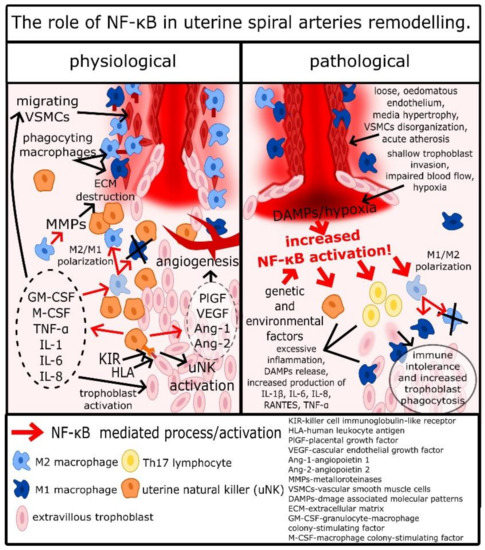
Figure 1. The role of NF-κB in uterine spiral arteries remodeling.
2.4. The Role of NF-κB in Trophoblast/uNK/Macrophage Crosstalk
Like it was described above, NF-κB has an important role from the very beginning of the pregnancy, as it regulates the expression of cytokines by epithelial cells, through the TGF-β1/TGF-βRs/PI3K/AKT/mTOR/IKK/NF-κB pathway [27][28][29]. Therefore, placentation starts with NF-κB regulated production of GM-CSF, M-CSF, IL-1α, IL-6, IL-8, LIF, RANTES, and MIP-1α by uterine epithelial cells. KIR receptors present on the surface of NKs have TLR-like properties, and Rajagopalan et al. show that association of KIR-2DL4 with HLA-G leads to activation of NF-κB [52][53]. Therefore, activation of uNKs by trophoblasts leads to the release of proinflammatory cytokines by uNKs in the NF-κB-dependent mechanism [54]. Decidual macrophages mostly have the M2 phenotype, and this type of cell has anti-inflammatory properties necessary for limitation of inflammation in uterus and regulation of spiral arteries remodeling [11]. Yunna et al. reported that macrophage polarization into the M2 phenotype is regulated by NF-κB, where increased NF-κB activation promotes M1 differentiation and decreased activation promotes M2 differentiation [55]. Chang et al. shed light on that process, describing that activation of TLR-2 leads to activation of p105/p65 heterodimers and p52/p65 increases the expression of p65 [56]. However, p65 in this mechanism, after transportation into the cytosol, is ubiquitinated and aggregated with other ubiquitinated p65 [56]. Such aggregates are internalized by autophagosomes and destructed [56]. Therefore, TLR2 activation in macrophages leads not to the activation of NF-κB, but to the decrease of p65 concentrations. According to Yu et al. among TLR2 ligands are hialuronian, endoplasmin, and high-mobility group protein 1 (HMGB1); therefore, the destruction of the extracellular matrix, cell apoptosis, and pyroptosis associated with spiral arteries remodeling and inflammation in the uterus can promote M2 polarization of decidual macrophages through decrease of NF-κB, which is possibly the inflammation self-restrain process, very important in regulation of implantation [57]. Regardless of the type of macrophages, NF-κB was reported to upregulate secretion of MMP-2 and MMP-9, which are crucial for disorganization of the extracellular matrix and trophoblasts invasion [14]. As was mentioned before, hypoxia occurs frequently during placentation and development of the placenta, and it stimulates angiogenesis. Hypoxia is an important NF-κB activator, and it was reported that in this mechanism, trophoblasts are stimulated to produce VEGFs, such as PlGF, which are critical for proper development of arteries in the placenta [14][58]. Interestingly, not only does the PlGF gene have NF-κB sites, but the endoglin gene and endoglin are also a part of the TGFβRs complex [55]. Finally, expression of many cytokines secreted by trophoblasts, uNKs, and macrophages, such as GM-CSF, M-CSF, TNF-α, IL-1α, IL-1β, IL-6, and IL-8, are regulated by NF-κB [1][13][14]. Therefore, NF-κB is a key mediator of placentation and spiral arteries remodeling, and disturbance of its regulatory function leads to a pathological development of pregnancy.
2.5. Abnormal Placentation in PE and the Role of NF-κB
During the physiological placentation, trophoblasts differentiate into villous cytotrophoblasts and syncytiotrophoblast, which form placenta villi and extravillous trophoblasts, which invade uterine decidua and take part in spiral arteries remodelling [17][18]. These extravillous trophoblasts infiltrate spiral arteries and replace VSMCs, forming wide, cone-like endings of these vessels, with a gentle widening. With progression of pregnancy, these trophoblasts are covered with endothelial cells, and such prepared vessels allow maternal blood to flow around placental capillaries and exchange oxygen, metabolites, etc., between mother and foetus [18]. Extravillous trophoblasts migrate against the blood flow, and some of them form a conglomerate in the proximal part of artery, resulting in transient hypoxia [17]. Hypoxia, alongside various cytokines, is an important mediator of spiral arteries remodeling, as was described in the section on trophoblast/uNK/macrophage crosstalk. It stimulates the invasion of extravillous trophoblasts into the arteries, but also helps to limit it and curb the extension of the remodeling to the distal end of the artery, as well as to slow down this process and make it steady [17][18].
Histological analysis of placenta samples acquired from women with PE revealed a significant impairment of spiral arteries remodeling, termed decidual vasculopathy [59][60]. Spiral arteries of women with PE were often characterized by loose, edematous endothelium, hypertrophy of the media, disorganization of the smooth muscle layer, and acute atherosis [59][61]. Stanek investigated placenta samples acquired from 230 women with early-onset PE, 261 women with late-onset PE, and 5059 healthy women [60]. Preeclampsia, regardless of the onset, was associated with increased occurrence of decidual arteriopathy, chronic hypoxic placental injury, villous infarction, membrane laminar necrosis, membrane microscopic chorionic pseudocysts, clusters of maternal floor multinucleated trophoblasts, excessive number of extravillous trophoblasts, and intervillous thrombi, in comparison to healthy individuals [60]. Chronic hypoxic placental injury was more common in early-onset PE than late-onset PE and correlated with more severe clinical outcomes [60] In PE, extravillous trophoblasts either infiltrate the uterine wall chaotically and deeper than normal or, as occurs more frequently, the invasion of trophoblasts is too shallow, limited only to the very end of arteries [14][17][18]. In this situation, there are very wide endings of arteries with significant narrowing right before them [17]. In such conditions, blood flows with greater pressure and mechanically widens aforementioned lacunas and also provides less oxygen than physiological, gently widening spiral arteries [62][14][17][18]. Moreover, stagnation of blood in lacunas leads to increased exposition of surrounding cell, inter alia trophoblasts, free radicals, harmful metabolites, cytokines, etc., and can lead to formation of clots [62][17][18]. This results in further, pathological intensification of inflammation, whilst hypoxia/reperfusion and excessive inflammation appear to be the main causes of the pathological remodeling [14][17][18]. To support that hypothesis, it should be mentioned that similar effects of hypoxia on blood vessels were observed in other tissues. For example, Kitchen et al. indicated that hypoxia leads to blood vessel remodeling in the blood–brain barrier, causing its function alteration and central nervous system oedema [63]. Therefore, it is very likely that the remodeling role of hypoxia is consistent throughout the human body, including the placenta.
In the case of PE, the aforementioned pathological inflammatory response is clearly mediated by NF-κB, as its abnormal elevation is characteristic for this disease [14]. High amounts of IL-1β, IL-6, IL-8, RANTES, and TNF-α, whose expression is regulated by NF-κB, leads to trophoblasts apoptosis, release of DAMPs, and further activation of NF-κB [1][14][17]. The aforementioned hypoxia/reperfusion insult leads to increased cell apoptosis and necrosis, release of DAMPs and PAMPs, and activation of NF-κB, and if these processes exceed abilities of decidual macrophages to phagocyte cell rests/DAMPs and “tide-up” the surrounding of spiral arteries, the remodeling process is disturbed [14][17]. Therefore, inflammation and NF-κB, which regulates production of proinflammatory cytokines and angiogenic factors, activation of macrophages, uNKs, and trophoblasts, are crucial for physiological placentation and spiral arteries remodeling; however, they must be carefully balanced through the pregnancy, as excessive activation of NF-κB leads to pathological placentation. There is still no clearly defined cause of the excessive activation of NF-κB and pathological inflammatory response in the uterus, although there are some interesting studies on this topic.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22020704
References
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866.
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132.
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. WIREs Syst. Biol. Med. 2016, 8, 227–241.
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the NF-κB signaling module. Oncogene 2006, 25, 6706–6716.
- Sakowicz, A.; Bralewska, M.; Pietrucha, T.; Habrowska-Górczyńska, D.E.; Piastowska-Ciesielska, A.W.; Gach, A.; Rybak-Krzyszkowska, M.; Witas, P.J.; Huras, H.; Grzesiak, M.; et al. Canonical, non-canonical and atypical pathways of nuclear factor κB activation in preeclampsia. Int. J. Mol. Sci. 2020, 21, 5574.
- Janssens, S.; Tschopp, J. Signals from within: The DNA-damage-induced NF-κB response. Cell Death Differ. 2006, 13, 773–784.
- Boisson, B.; Puel, A.; Picard, C.; Casanova, J.-L. Human IκBα Gain of Function: A Severe and Syndromic Immunodeficiency. J. Clin. Immunol. 2017, 37, 397–412.
- Cooke, E.L.; Uings, I.J.; Xia, C.L.; Woo, P.; Ray, K.P. Functional analysis of the interleukin-1-receptor-associated kinase (IRAK-1) in interleukin-1β-stimulated nuclear factor κB (NF-κB)pathway activation: IRAK-1 associates with the NF-κB essential modulator (NEMO) upon receptor stimulation. Biochem. J. 2001, 359, 403–410.
- Liu, G.; Park, Y.-J.; Abraham, E. Interleukin-1 receptor-associated kinase (IRAK)-1-mediated NF-κ activation requires cytosolic and nuclear activity. FASEB J. 2008, 22, 2285–2296.
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85.
- Mor, G.; Aldo, P.; Alvero, A.B. The unique immunological and microbial aspects of pregnancy. Nat. Rev. Immunol. 2017, 17, 469–482.
- Nakamura, H.; Kimura, T.; Ogita, K.; Koyama, S.; Tsujie, T.; Tsutsui, T.; Shimoya, K.; Koyama, M.; Kaneda, Y.; Murata, Y. Alteration of the timing of implantation by in vivo gene transfer: Delay of implantation by suppression of nuclear factor κB activity and partial rescue by leukemia inhibitory factor. Biochem. Biophys. Res. Commun. 2004, 321, 886–892.
- Sakowicz, A. The role of NFκB in the three stages of pregnancy—Implantation, maintenance, and labour: A review article. BJOG Int. J. Obstet. Gynaecol. 2018, 125, 1379–1387.
- Armistead, B.; Kadam, L.; Drewlo, S.; Kohan-Ghadr, H.-R. The Role of NFκB in Healthy and Preeclamptic Placenta: Trophoblasts in the Spotlight. Int. J. Mol. Sci. 2020, 21, 1775.
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and pregnancy: The role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87.
- Malinen, M.; Niskanen, E.A.; Kaikkonen, M.U.; Palvimo, J.J. Crosstalk between androgen and pro-inflammatory signaling remodels androgen receptor and NF-κB cistrome to reprogram the prostate cancer cell transcriptome. Nucleic Acids Res. 2017, 45, 619–630.
- James, J.L.; Whitley, G.S.; Cartwright, J.E. Pre-eclampsia: Fitting together the placental, immune and cardiovascular pieces. J. Pathol. 2010, 221, 363–378.
- Soares, M.J.; Varberg, K.M.; Iqbal, K. Hemochorial placentation: Development, function, and adaptations. Biol. Reprod. 2018, 99, 196–211.
- Bromfield, J.J. Seminal fluid and reproduction: Much more than previously thought. J. Assist. Reprod. Genet. 2014, 31, 627–636.
- Robertson, S.A.; Mau, V.J.; Hudson, S.N.; Tremellen, K.P. Cytokine-Leukocyte Networks and the Establishment of Pregnancy. Am. J. Reprod. Immunol. 1997, 37, 438–442.
- Schjenken, J.E.; Robertson, S.A. Seminal fluid and immune adaptation for pregnancy—Comparative biology in mammalian species. Reprod. Domest. Anim. 2014, 49, 27–36.
- Watson, J.G.; Carroll, J.; Chaykin, S. Reproduction in mice: The fate of spermatozoa not involved in fertilization. Gamete Res. 1983, 7, 75–84.
- Carp, H.; Serr, D.M.; Mashiach, S.; Nebel, L. Influence of insemination on the implantation of transferred rat blastocysts. Gynecol. Obstet. Investig. 1984, 18, 194–198.
- Tremellen, K.P.; Valbuena, D.; Landeras, J.; Ballesteros, A.; Martinez, J.; Mendoza, S.; Norman, R.J.; Robertson, S.A.; Simón, C. The effect of intercourse on pregnancy rates during assisted human reproduction. Hum. Reprod. 2000, 15, 2653–2658.
- Maxwell, W.M.C.; Evans, G.; Mortimer, S.T.; Gillan, L.; Gellatly, E.S.; McPhie, C.A. Normal fertility in ewes after cervical insemination with frozen-thawed spermatozoa supplemented with seminal plasma. Reprod. Fertil. Dev. 1999, 11, 123–126.
- Redman, C.W.G.; Sargent, I.L. Immunology of pre-eclampsia. Am. J. Reprod. Immunol. 2010, 63, 534–543.
- Torrealba, N.; Rodríguez-Berriguete, G.; Fraile, B.; Olmedilla, G.; Martínez-Onsurbe, P.; Guil-Cid, M.; Paniagua, R.; Royuela, M. Expression of several cytokines in prostate cancer: Correlation with clinical variables of patients. Relationship with biochemical progression of the malignance. Cytokine 2017, 89, 105–115.
- Krstić, J.; Trivanović, D.; Mojsilović, S.; Santibanez, J.F. Transforming growth factor-beta and oxidative stress interplay: Implications in tumorigenesis and cancer progression. Oxid. Med. Cell. Longev. 2015, 2015.
- Torrealba, N.; Vera, R.; Fraile, B.; Martínez-Onsurbe, P.; Paniagua, R.; Royuela, M. TGF-β/PI3K/AKT/mTOR/NF-kB pathway. Clinicopathological features in prostate cancer. Aging Male 2019.
- Daimon, E.; Wada, Y. Role of neutrophils in matrix metalloproteinase activity in the preimplantation mouse uterus. Biol. Reprod. 2005, 73, 163–171.
- Chow, P.H.; Jiang, H.Y.; Poon, H.K.; Lee, K.H.; O, W.S. Embryos sired by males without accessory sex glands induce failure of uterine support: A study of VEGF, MMP and TGF expression in the golden hamster. Anat. Embryol. 2003, 206, 203–213.
- Yang, H.L.; Thiyagarajan, V.; Shen, P.C.; Mathew, D.C.; Lin, K.Y.; Liao, J.W.; Hseu, Y.C. Anti-EMT properties of CoQ0 attributed to PI3K/AKT/NFKB/MMP-9 signaling pathway through ROS-mediated apoptosis. J. Exp. Clin. Cancer Res. 2019, 38, 186.
- Faas, M.M.; de Vos, P. Uterine NK cells and macrophages in pregnancy. Placenta 2017, 56, 44–52.
- Harris, L.K. Review: Trophoblast-Vascular Cell Interactions in Early Pregnancy: How to Remodel a Vessel. Placenta 2010, 31.
- Sojka, D.K.; Yang, L.; Yokoyama, W.M. Uterine Natural Killer Cells. Front. Immunol. 2019, 10, 960.
- Zhang, J.; Tian, Z. UNK cells: Their role in tissue re-modelling and preeclampsia. Semin. Immunopathol. 2007, 29, 123–133.
- Liu, W.; Luo, M.; Zou, L.; Liu, X.; Wang, R.; Tao, H.; Wu, D.; Zhang, W.; Luo, Q.; Zhao, Y. uNK cell-derived TGF-β1 regulates the long noncoding RNA MEG3 to control vascular smooth muscle cell migration and apoptosis in spiral artery remodeling. J. Cell. Biochem. 2019, 120, 15997–16007.
- Trundley, A.; Moffett, A. Human uterine leukocytes and pregnancy. Tissue Antigens 2004, 63, 1–12.
- Chakraborty, D.; Rumi, M.A.K.; Konno, T.; Soares, M.J. Natural killer cells direct hemochorial placentation by regulating hypoxia-inducible factor dependent trophoblast lineage decisions. Proc. Natl. Acad. Sci. USA 2011, 108, 16295–16300.
- Naruse, K.; Lash, G.E.; Innes, B.A.; Otun, H.A.; Searle, R.F.; Robson, S.C.; Bulmer, J.N. Localization of matrix metalloproteinase (MMP)-2, MMP-9 and tissue inhibitors for MMPs (TIMPs) in uterine natural killer cells in early human pregnancy. Hum. Reprod. 2009, 24, 553–561.
- Chen, J.; Khalil, R.A. Matrix Metalloproteinases in Normal Pregnancy and Preeclampsia. Prog. Mol. Biol. Transl. Sci. 2017, 148, 87–165.
- Lash, G.E.; Pitman, H.; Morgan, H.L.; Innes, B.A.; Agwu, C.N.; Bulmer, J.N. Decidual macrophages: Key regulators of vascular remodeling in human pregnancy. J. Leukoc. Biol. 2016, 100, 315–325.
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; DeNardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288.
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediators Inflamm. 2015, 2015, 816460.
- Brown, M.B.; von Chamier, M.; Allam, A.B.; Reyes, L. M1/M2 macrophage polarity in normal and complicated pregnancy. Front. Immunol. 2014, 5, 606.
- Houser, B.L. Decidual macrophages and their roles at the maternal-fetal interface. Yale J. Biol. Med. 2012, 85, 105–118.
- Qi, X.F.; Kim, D.H.; Yoon, Y.S.; Jin, D.; Huang, X.Z.; Li, J.H.; Deung, Y.K.; Lee, K.J. Essential involvement of cross-talk between IFN-γ and TNF-α in CXCL10 production in human THP-1 monocytes. J. Cell. Physiol. 2009, 220, 690–697.
- Xia, J.B.; Liu, G.H.; Chen, Z.Y.; Mao, C.Z.; Zhou, D.C.; Wu, H.Y.; Park, K.S.; Zhao, H.; Kim, S.K.; Cai, D.Q.; et al. Hypoxia/ischemia promotes CXCL10 expression in cardiac microvascular endothelial cells by NFkB activation. Cytokine 2016, 81, 63–70.
- Zhao, H.; Kalish, F.S.; Wong, R.J.; Stevenson, D.K. Hypoxia regulates placental angiogenesis via alternatively activated macrophages. Am. J. Reprod. Immunol. 2018, 80, e12989.
- Dominguez, F.; Martínez, S.; Quiñonero, A.; Loro, F.; Horcajadas, J.A.; Pellicer, A.; Simón, C. CXCL10 and IL-6 induce chemotaxis in human trophoblast cell lines. Mol. Hum. Reprod. 2008, 14, 423–430.
- Plaks, V.; Birnberg, T.; Berkutzki, T.; Sela, S.; BenYashar, A.; Kalchenko, V.; Mor, G.; Keshet, E.; Dekel, N.; Neeman, M.; et al. Uterine DCs are crucial for decidua formation during embryo implantation in mice. J. Clin. Investig. 2008, 118, 3954–3965.
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling Natural Killer Cell Responses: Integration of Signals for Activation and Inhibition. Annu. Rev. Immunol. 2013, 31, 227–258.
- Rajagopalan, S.; Moyle, M.W.; Joosten, I.; Long, E.O. DNA-PKcs controls an endosomal signaling pathway for a proinflammatory response by natural killer cells. Sci. Signal. 2010, 3.
- Rajasekaran, K.; Kumar, P.; Schuldt, K.M.; Peterson, E.J.; Vanhaesebroeck, B.; Dixit, V.; Thakar, M.S.; Malarkannan, S. Signaling by Fyn-ADAP via the Carma1-Bcl-10-MAP3K7 signalosome exclusively regulates inflammatory cytokine production in NK cells. Nat. Immunol. 2013, 14, 1127–1136.
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090.
- Chang, C.P.; Su, Y.C.; Lee, P.H.; Lei, H.Y. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy 2013, 9, 619–621.
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603.
- Chau, K.; Hennessy, A.; Makris, A. Placental growth factor and pre-eclampsia. J. Hum. Hypertens. 2017, 31, 782–786.
- Hecht, J.L.; Zsengeller, Z.K.; Spiel, M.; Karumanchi, S.A.; Rosen, S. Revisiting decidual vasculopathy. Placenta 2016, 42, 37–43.
- Stanek, J. Histological Features of Shallow Placental Implantation Unify Early-Onset and Late-Onset Preeclampsia. Pediatr. Dev. Pathol. 2019, 22, 112–122.
- Kim, J.-Y.; Kim, Y.M. Acute Atherosis of the Uterine Spiral Arteries: Clinicopathologic Implications. J. Pathol. Transl. Med. 2015, 49, 462–471.
- Ahmed, A.; Rezai, H.; Broadway-Stringer, S. Evidence-Based Revised View of the Pathophysiology of Preeclampsia. Adv. Exp. Med. Biol. 2017, 956, 355–374.
- Kitchen, P.; Salman, M.M.; Halsey, A.M.; Clarke-Bland, C.; MacDonald, J.A.; Ishida, H.; Vogel, H.J.; Almutiri, S.; Logan, A.; Kreida, S.; et al. Targeting Aquaporin-4 Subcellular Localization to Treat Central Nervous System Edema. Cell 2020, 181, 784–799.