Vitamin C is implicated in various bodily functions due to its unique properties in redox homeostasis. Moreover, vitamin C also plays a great role in restoring the activity of 2-oxoglutarate and Fe2+ dependent dioxygenases (2-OGDD), which are involved in active DNA demethylation (TET proteins), the demethylation of histones, and hypoxia processes. Therefore, vitamin C may be engaged in the regulation of gene expression or in a hypoxic state.
- vitamin C
- SVCT polymorphisms
- carcinogenesis
1. Vitamin C Properties and Redox Homeostasis
Vitamin C, a common name for l-ascorbic acid or l-ascorbate, is a six-carbon lactone. It can be synthesized from glucose by enzyme l-gulono-1,4-lactone oxidase, which can be found in almost every mammal’s liver, with the exception of humans, other primates, bats, and guinea pigs [1][2]. Consequently, these species must provide their organisms with l-ascorbate exogenously. Since l-ascorbate is a key factor in intensifying the activity of numerous enzymes located throughout the human body, the external intake of vitamin C is particularly important. Its unique properties are connected with its structure and biochemistry: ascorbic acid can be transformed into ascorbate monoanion or ascorbate dianion via the dissociation of one or two hydrogen ions from hydroxyl groups located at carbon 2 and carbon 3. In physiological pH ascorbate functions as a monoanion, which is its primary form. Ascorbic acid is known to be a significant reducing factor, easily subject to the compilations of two one-electron oxidations. The first one is accomplished via ascorbate radical generation [3], providing a specific chemical structure (resonance stabilization) with a stable yet not highly reactive form [4]. The ascorbate radical can then undergo the second one-electron oxidation to form dehydroascorbic acid (DHA) (Figure 1) [3]. This ability makes ascorbate a particularly good defender against other free radicals by substituting them with more stable and less reactive compounds [4]. Furthermore, in vitro studies demonstrated that an increased level of ascorbate can act as a stimulus to cooperate with superoxide dismutase in the removal of superoxides [5]. DHA and ascorbate radicals can also be reduced to ascorbate in a reversible manner [3].
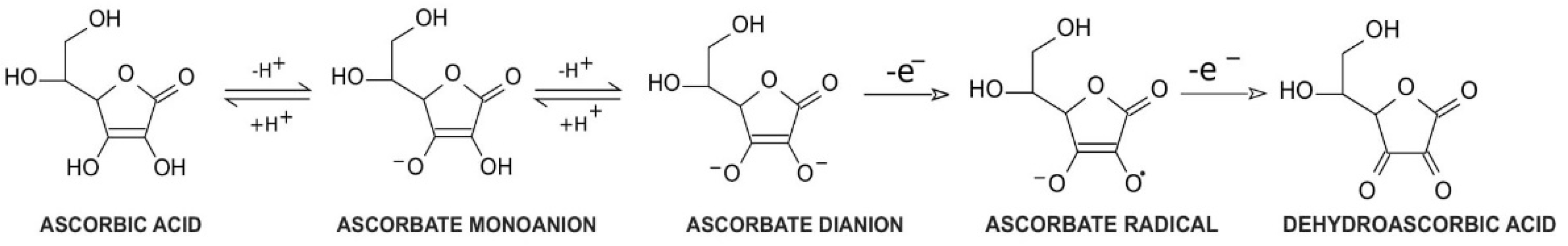
Figure 1. Vitamin C redox states. Ascorbic acid undergoes two reversible hydrogen dissociations to form ascorbate monoanions and ascorbate dianions, respectively. Then, ascorbate dianons can be subsequently subjected to one-electron oxidation to create ascorbate radicals. These are not highly reactive components; however, they can undergo another one-electron oxidation to form dehydroascorbic acid.
Despite its ability to act as a reducing agent, ascorbate can also perform a key role as an oxidative factor. Although the oxidation process of ascorbate occurs mainly in the presence of catalytic metals, the process can be performed by ascorbate itself (autooxidation) but at a significantly slower rate in neutral pH [6]. The effect of vitamin C oxidation is hydrogen peroxide formation, which can affect cellular metabolism, by changing intracellular redox stability [6][7]. Moreover, vitamin C can act as a prodrug through its ability to serve as a reducing and oxidizing factor [7]. The privileged form of vitamin C is determined by the vitamin C concentration in plasma. In physiological concentrations, ascorbic acid preferentially exerts its antioxidant functions, which help restore cellular 2-oxoglutarate and Fe2+ dependent dioxygenase activity (2-OGDDs) [8]. On the other hand, a higher level of ascorbic acid is linked to pro-oxidative functions [7].
2. Vitamin C and Its Role in 2-OGDD Enzymes Activity
Due to its antioxidant potential, vitamin C can also serve as an enzyme cofactor. 2-OGDD enzymes require 2-oxoglutarate (2-OG) and Fe2+ to maintain their catalytic activity. However, most 2-OGGDs also require ascorbate as a cofactor [9]. The family of 2-OGDDs consists of more than 60 enzymes that contain specific double-stranded β-helix (DSBH) motifs with Fe2+ ions in their cores. Their catalytic functions relate to the hydroxylation of particular substrates with the decarboxylation of 2-OG to succinate in the presence of oxygen [10]. Ascorbic acid crucially increases the rate of the reaction catalyzed by 2-OGGD by targeting its catalytic domain and regenerating iron ions from Fe3+ to Fe2+ (Figure 2) [11].
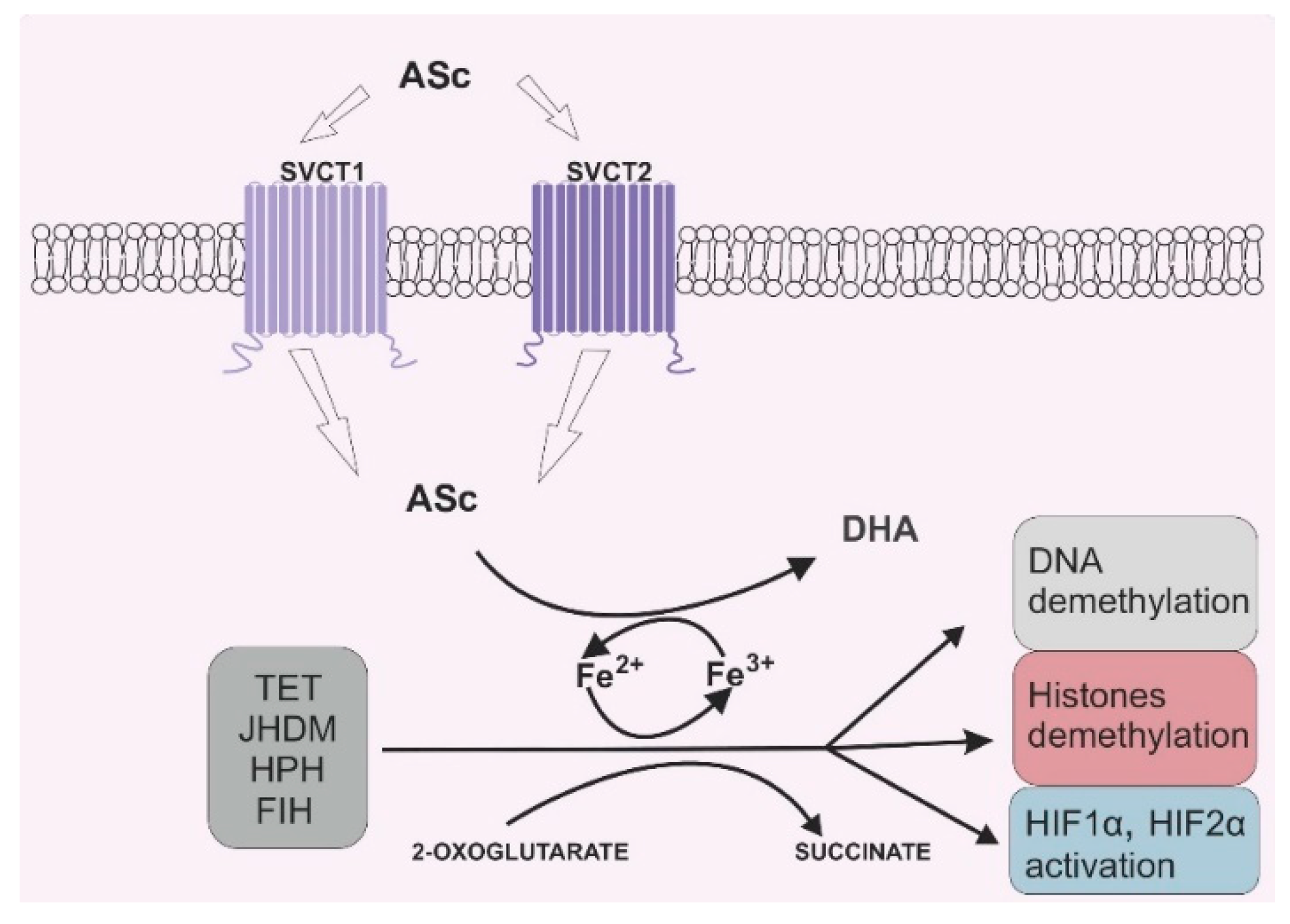
Figure 2. Vitamin C’s role in 2-oxoglutarate and Fe2+ dependent dioxygenase (2-OGDD) activity. Legend abbreviations: ASc—ascorbate; SCVT1—sodium dependent vitamin C transporter 1; SCVT2—sodium dependent vitamin C transporter 2; DHA—dehydroascorbic acid, TET—ten–eleven translocation proteins; JHDM—Jumonji C-domain-containing histone demethylases; HPH—hypoxia-inducible factor prolyl hydroxylases; FIH—factor-inhibiting hypoxia-inducible factor; HIF1α—subunit α of the hypoxia inducible factor 1; HIF2 α—subunit α of the hypoxia inducible factor 2. Ascorbate can be engaged in restoring the activity of 2-OGDD enzymes by reducing Fe3+ to Fe2+, which can be found in the catalytic center of those enzymes. 2-OGDD enzymes also require 2-oxoglutarate to maintain their catalytic activity. TET, JHDM, HPH and FIH are members of the 2-OGDD family, and their main roles in cell biochemistry are based on DNA demethylation (TET proteins), histone demethylation (JHDM proteins) and HIF1α and HIF2 α activation (HPH and FIH proteins). A more detailed description is given in the text.
Research over the past decade has highlighted that vitamin C has a crucial function in epigenetics. In 2009, Tahiliani et al. shed new light on the epigenetic field. The authors discovered that ten-eleven-translocation enzymes (TET: TET1, TET2, TET3), originally considered as chromosomal translocations of the genes MLL and LCX (t(10;11)(q22;q23)) in acute myeloid leukemia patients [12][13], are actually responsible for changing the DNA methylation pattern [14]. Their research indicated that TETs have the ability to induce the hydroxylation of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), which leads to DNA demethylation [14]. Later developments in this field revealed that TETs are also involved in the further hydroxylation of 5-hmC to the other cytosine derivatives, which can be subsequently excised by DNA glycosylases [15]. A number of studies have found that ascorbate is an important compound for TET protein activity, since these proteins belong to the 2-OGDD superfamily. It has been proven that ascorbic acid enhances 5-hmC generation by promoting the hydroxylation of 5-mC provided by TET proteins [16]. Additionally, it was conclusively demonstrated that ascorbic acid’s impact on TET proteins is not only due to its reducing ability, as other strong oxidizing agents do not present a similar activity [17]. In line with this finding, vitamin C was proposed to be an agent that promotes DNA demethylation [16][17]. DNA methylation and demethylation are not only linked with cytosine modifications, but also with chromatin reorganization through changes in the amino acids in histones. The enzymes responsible for histone demethylation are Jumonji C-domain-containing histone demethylases (JHDMs), which also belong to the 2-OGDD superfamily [18]. As mentioned in the literature review, more than 20 JHDMs are capable of eliminating the methyl groups of lysines, which are located in histones [9][19]. Previous studies identified that the methylation of lysines can induce or inhibit transcription depending on their location in the histones. Methylation located at lysine 9 in histone H3 and at lysine 20 in histone H4 provides gene silencing, whereas the methylation of lysine 4, lysine 36, and lysine 79 in histone H3 is linked with enhancing transcription [20][21]. A large and growing body of evidence has investigated the essential role of the impaired balance between methylation and demethylation during cancer initiation and progression [22][23][24][25]. In recent years, there has been an increasing interest in TET or JHDM mutations in different cancers, especially hematological ones [26][27][28]. Therefore, the plausible effects of vitamin C in cancer therapy have been the central aim of numerous works of research.
Other members of the 2-OGDD family are the hypoxia-inducible factors prolyl hydroxylases (HPHs) and the factor-inhibiting hypoxia-inducible factor (FIH), which are arranged during the initiation of the degradation of subunit α of the hypoxia inducible factors HIF1 and HIF2. HIFs take part in cellular adaptation to an anaerobic state, which frequently occurs in tumors [29]. The tumor microenvironment is distinct from that observed in healthy tissues. Tumor cells have the ability to divide very quickly and frequently. Thus, boosting the tumor mass generates separation from blood vessels. Although angiogenesis is common in cancer development, the newly formed vessels are distinct from healthy ones in terms of their structures and functions [30]. Taken together, there is a decrease in the partial pressure of oxygen in tumor masses compared to the pressure observed in corresponding healthy tissue [31][32]. A hypoxic state in tumors can increase the tumor’s invasiveness and ability to metastasize [33]. HIFs are composed of two constitutively expressed subunits, α and β. In normoxic conditions, subunit α is hydroxylated by HPH and FIH; after a multistage post-translational process, subunit α is degraded by proteasomes [29]. However, in a hypoxic state, subunit α is stabilized, followed by bonding with the β subunit, which eventually facilitates the transcription of specific genes. HIF1-α and HIF2-α differ from each other via transcription regulation: HIF1-α is responsible mainly for the regulation of genes involved in cellular metabolic changes, and HIF2-α is responsible for the control of genes involved in cellular signaling and extracellular matrix remodeling factors [34][35][36]. Moreover, both factors exhibit different expressions during hypoxia: in an acute hypoxic state, HIF1-α is highly expressed, whereas long-termed hypoxia results in HIF2-α accumulation [37]. Given that subunit α stabilization is crucial for the expression of HIFs, HPHs and FIH are major factors contributing to cell adaptation under an anaerobic state. Hence, any dysregulation of these enzymes may itself be a trigger of cancer initiation.
3. Effects of Vitamin C on Cancer Cells
Due to the pleiotropic functions of vitamin C, the idea of using this compound as a potential anti-cancer drug is not new. However, the past decade has seen increasingly rapid advances in novel approaches to cancer therapy. There is a general agreement that cancer patients exhibit significantly lower level of vitamin C in their plasma compared to healthy subjects [8][38][39]. Moreover, previous studies conclusively demonstrated that ascorbate deficiency can improve the invasiveness of cancer cells [40][41]. One plausible explanation of vitamin C depletion is oxidative stress in cells and reactive oxygen species (ROS) formation over the course of carcinogenesis. It is widely known that cancer cells exhibit different metabolic processes as a result of the dysfunction of the cellular organelles (mainly the mitochondria). This eventually leads to excessive ROS generation, followed by chronic inflammation [42][43]. Vitamin C acts as a radical scavenger due to its antioxidant ability and plays a pivotal role in ROS elimination, so vitamin C levels may eventually decline when free radicals are excessively generated [44]. Therefore, it was suggested that vitamin C may have great value in cancer therapy. In line with this hypothesis, a considerable amount of literature has been published on vitamin C’s ability to destroy cancer cells in vitro and in vivo, even under pharmacological concentrations [7][45][46]. Moreover, it was also indicated that ascorbate has the ability to inhibit cancer growth [47]. There are several possible ways that vitamin C can exert anti-cancer action. One of them is vitamin C’s pro-oxidant ability, which is revealed mainly at high doses and can be accessed only via intravenous intake. Several studies provided by Chen et al. showed that vitamin C may produce hydrogen peroxide (H2O2) as an intermediate of ascorbate radical generation in cancer cells [7][45][47]. H2O2, as one of ROS, plays a key role in maintaining the cellular redox state, it may have an impact on disrupting cancer cells’ metabolism [48]. According to Uetaki et al., the main target for vitamin C’s anti-cancer action is the inhibition of glycolysis via a decrease in NAD [48]. Interestingly, vitamin C cytotoxicity is observed only in tumor cells while omitting normal ones [45]. This difference may be the result of an altered mode of ATP generation in cancer cells: instead of oxidative proliferation, such cells preferentially undergo glycolysis, even in an aerobic state. This process is commonly known as the Warburg effect [49]. Hence, vitamin C is targeted in the process and is mainly responsible for energy production in the tumor cells. Furthermore, some cancer-specific mutations are actually associated with the Warburg effect, as previously demonstrated [50][51]. KRAS or BRAF mutations that occur in colorectal cancer may also contribute to glucose uptake and GLUT1 overexpression [52]. As mentioned, GLUT1 transports DHA, which is reduced to ascorbic acid directly after transferring to the cell [53]. A great amount of GLUT1 in KRAS and BRAF mutant cells can be a target for high doses of vitamin C, which can ultimately lead to ROS generation and cancer cell death [51].
However, abnormal cell functions during carcinogenesis are predisposed to reduced oxygen availability [54], and a hypoxic state in cancer cells may restrain the cytotoxic effect of vitamin C [55]. Therefore, there is a second explanation for the selective attack of vitamin C on cancer cells based on vitamin C’s potential to react with iron ions, which can be found in the catalytic centers of multiple enzymes [3]. According to the hypothesis proposed by Ngo et al., there is an abundance of Fe2+ ions in specific cancer microenvironments, which can generate H2O2 and •OH through a reaction with vitamin C [56]. Moreover, the enrichment of Fe2+ inside cancer cells may stimulate the diffusion of H2O2 from extracellular space, which is an intermediate for vitamin C autooxidation [56]. In both cases, an excessive amount of H2O2 may lead to cytotoxicity in cancer cells.
A large and growing body of studies have demonstrated that the balance between methylation and demethylation is disturbed in many types of cancers [23][57][58][59]. It was found that 5-mC changes involve 5-hmC reductions in tumors [23][24]. Such alterations are proven to have an impact on the transcription of key genes in the human body, including oncogenes and tumor suppressor genes, which may provoke carcinogenesis [60]. Moreover, it was recently detected that the vitamin C level correlates with 5-hmC [61] and, since 5-hmC is potentially involved in the regulation of gene expression [25], ascorbate may also be involved in this process. Ascorbate is a co-factor of TET proteins, which are members of the 2-OGGD family. Several in vitro [16][62][63][64] and in vivo [65] studies have established that ascorbate has the ability to increase 5-hmC. It is also noteworthy that vitamin C removal in vitro correlates with a remarkable decrease in the 5-hmC level with a parallel increase in the 5-mC level [62]. Furthermore, TET2 mutations often occur in hematological malignancies, with approximately 10% in acute myeloid leukemia, 30% in myelodysplastic syndrome, and 50% in chronic myelomonocytic leukemia [66]. Notably, in the case of TET2 mutations, application of vitamin C has the ability to restore TET2 deficiency and promote DNA demethylation [67]. The relationship between TET2 enzymes and ascorbate therapy in leukemic cells was also detected in a study by Zhao et al. [68], who found that vitamin C administration increased TET2 activity. However, in terms of TET2 expression, a similar effect was not detected [68]. Vitamin C in cancer therapy has other beneficial effects on the suppression of tumor growth and metastasis (discussed below) [63][69][70][71], which may be associated with changes in the transcription and expression of genes caused by changes in DNA methylation. As mentioned above, other enzymes regulating the epigenome that depend upon vitamin C are JHDMs connected with chromatin changes [18]. Several studies have identified vitamin C as a crucial agent contributing to the JHDM-dependent chromatin demethylation that occurs during hematopoiesis and somatic cell reprogramming [72][73][74][75]. Given that DNA methylation changes are the most prevalent among the various epigenetic processes, vitamin C likely has great value in the regulation of gene expression.
4. Vitamin C in Adjuvant Cancer Therapy
Vitamin C has been frequently prescribed as a complementary or alternative treatment for cancer patients in previous decades. Following the report that intravenous ascorbic acid administration is safe [76], there has been an increasing amount of literature on implementing ascorbic acid therapy in various cancers. However, one question that needs to be asked is whether vitamin C influences conventional chemotherapy or radiotherapy. Vitamin C seems to be effective in preventing the side effects of chemotherapy and radiotherapy [77][78]. However, the ROS generated during radiation may be eliminated by vitamin C, which can diminish the therapeutic effect of radiotherapy. Similarly, chemotherapeutic agents whose pharmacological actions are based on ROS generation may also be inefficient under vitamin C supplementation [79]. Therefore, there is still a need for further research in this field.
This entry is adapted from the peer-reviewed paper 10.3390/nu12123869
References
- Nishikimi, M.; Yagi, K. Biochemistry and molecular biology of ascorbic acid biosynthesis. Subcell. Biochem. 1996, 25, 17–39, doi:10.1007/978-1-4613-0325-1_2.
- Cui, J.; Pan, Y.-H.; Zhang, Y.; Jones, G.; Zhang, S. Progressive pseudogenization: Vitamin C synthesis and its loss in bats. Mol. Biol. Evol. 2010, 28, 1025–1031, doi:10.1093/molbev/msq286.
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457, doi:10.1016/j.bbcan.2012.06.003.
- Bielski, B.H.J. Chemistry of ascorbic acid radicals. In Ascorbic Acid: Chemistry, Metabolism, and Uses; Advances in Chemistry; American Chemical Society: Washington, WA, USA: 1982; Volume 200, pp. 81–100, ISBN 978-0-8412-0632-8
- Jackson, T.S.; Xu, A.; Vita, J.A.; Keaneyjr, J.F. Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circ. Res. 1998, 83, 916–922, doi:10.1161/01.res.83.9.916.
- Buettner, G.R. In the absence of catalytic metals ascorbate does not autoxidize at pH 7: Ascorbate as a test for catalytic metals. J. Biochem. Biophys. Methods 1988, 16, 27–40, doi:10.1016/0165-022x(88)90100-5.
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754, doi:10.1073/pnas.0702854104.
- Liu, M.; Ohtani, H.; Zhou, W.; Ørskov, A.D.; Charlet, J.; Zhang, Y.W.; Shen, H.; Baylin, S.B.; Liang, G.; Grønbæk, K.; et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc. Natl. Acad. Sci. USA 2016, 113, 10238–10244, doi:10.1073/pnas.1612262113.
- Clifton, I.J.; McDonough, M.A.; Ehrismann, D.; Kershaw, N.J.; Granatino, N.; Schofield, C.J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem. 2006, 100, 644–669, doi:10.1016/j.jinorgbio.2006.01.024.
- A McDonough, M.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672, doi:10.1016/j.sbi.2010.08.006.
- Gorres, K.L.; Raines, R.T. Prolyl 4-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 106–124, doi:10.3109/10409231003627991.
- Ono, R.; Taki, T.; Taketani, T.; Taniwaki, M.; Kobayashi, H.; Hayashi, Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10;11)(q22;q23). Cancer Res. 2002, 62, 4075–4080.
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637–641, doi:10.1038/sj.leu.2402834.
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935, doi:10.1126/science.1170116.
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303, doi:10.1126/science.1210597.
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate Induces Ten-Eleven Translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-Hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674, doi:10.1074/jbc.c113.464800.
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances tet-mediated 5-Methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403, doi:10.1021/ja4028346.
- Tsukada, Y.-I.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nat. Cell Biol. 2006, 439, 811–816, doi:10.1038/nature04433.
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727, doi:10.1038/nrg1945.
- Miller, J.L.; Grant, P.A. The role of DNA methylation and histone modifications in transcriptional regulation in humans. Subcell. Biochem. 2013, 61, 289–317, doi:10.1007/978-94-007-4525-4_13.
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849, doi:10.1038/nrm1761.
- Clark, S.; Melki, J. DNA methylation and gene silencing in cancer: Which is the guilty party? Oncogene 2002, 21, 5380–5387, doi:10.1038/sj.onc.1205598.
- Haffner, M.C.; Chaux, A.; Meeker, A.K.; Esopi, D.M.; Gerber, J.; Pellakuru, L.G.; Toubaji, A.; Argani, P.; Iacobuzio-Donahue, C.; Nelson, W.G.; et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2011, 2, 627–637, doi:10.18632/oncotarget.316.
- Kudo, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Asaoka, Y.; Ijichi, H.; Nagae, G.; Yoshida, H.; Aburatani, H.; Koike, K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012, 103, 670–676, doi:10.1111/j.1349-7006.2012.02213.x.
- Yildirim, O.; Li, R.; Hung, J.-H.; Chen, P.B.; Dong, X.; Ee, L.-S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-Hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510, doi:10.1016/j.cell.2011.11.054.
- Andricovich, J.; Kai, Y.; Tzatsos, A. Lysine-specific histone demethylases in normal and malignant hematopoiesis. Exp. Hematol. 2016, 44, 778–782, doi:10.1016/j.exphem.2016.05.006.
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation inTET2in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301, doi:10.1056/nejmoa0810069.
- Kosmider, O.; Gelsi-Boyer, V.; Ciudad, M.; Racoeur, C.; Jooste, V.; Vey, N.; Quesnel, B.; Fenaux, P.; Bastie, J.-N.; Beyne-Rauzy, O.; et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009, 94, 1676–1681, doi:10.3324/haematol.2009.011205.
- Brahimi-Horn, M.C.; Pouysségur, J. HIF at a glance. J. Cell Sci. 2009, 122, 1055–1057, doi:10.1242/jcs.035022.
- Postovit, L.-M.; Abbott, D.E.; Payne, S.L.; Wheaton, W.W.; Margaryan, N.V.; Sullivan, R.; Jansen, M.K.; Csiszar, K.; Hendrix, M.J.; Kirschmann, D.A. Hypoxia/reoxygenation: A dynamic regulator of lysyl oxidase-facilitated breast cancer migration. J. Cell. Biochem. 2008, 103, 1369–1378, doi:10.1002/jcb.21517.
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1236, doi:10.1089/ars.2007.1628.
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253, doi:10.1111/j.1582-4934.2011.01258.x.
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47, doi:10.1038/nrc704.
- Semenza, G.L. Signal transduction to hypoxia-inducible factor 1. Biochem. Pharmacol. 2002, 64, 993–998, doi:10.1016/s0006-2952(02)01168-1.
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684, doi:10.1038/nm0603-677
- Downes, N.L.; Laham-Karam, N.; Kaikkonen-Määttä, M.; Ylä-Herttuala, S. Differential but complementary HIF1α and HIF2α transcriptional regulation. Mol. Ther. 2018, 26, 1735–1745, doi:10.1016/j.ymthe.2018.05.004.
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423, doi:10.1016/j.ccr.2006.08.026.
- Mayland, C.R.; Bennett, M.I.; Allan, K. Vitamin C deficiency in cancer patients. Palliat. Med. 2005, 19, 17–20, doi:10.1191/0269216305pm970oa.
- Choi, M.-A.; Kim, B.-S.; Yu, R. Serum antioxidative vitamin levels and lipid peroxidation in gastric carcinoma patients. Cancer Lett. 1999, 136, 89–93, doi:10.1016/s0304-3835(98)00312-7.
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp. Oncol. 2011, 33, 226–230.
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int. J. Oncol. 2013, 42, 55–64, doi:10.3892/ijo.2012.1712.
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322, doi:10.1089/ars.2011.4414.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462, doi:10.1016/j.cub.2014.03.034.
- Galloway, S.P.; McMillan, D.C.; Sattar, N. Effect of the inflammatory response on trace element and vitamin status. Ann. Clin. Biochem. 2000, 37, 289–297, doi:10.1258/0004563001899429.
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609, doi:10.1073/pnas.0506390102.
- Takemura, Y.; Satoh, M.; Satoh, K.; Hamada, H.; Sekido, Y.; Kubota, S. High dose of ascorbic acid induces cell death in mesothelioma cells. Biochem. Biophys. Res. Commun. 2010, 394, 249–253, doi:10.1016/j.bbrc.2010.02.012.
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109, doi:10.1073/pnas.0804226105.
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 2015, 5, 13896, doi:10.1038/srep13896.
- Otto, A.M. Warburg effect(s)—A biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab. 2016, 4, 1–8, doi:10.1186/s40170-016-0145-9.
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.V.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559, doi:10.1126/science.1174229.
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.D.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396, doi:10.1126/science.aaa5004.
- Kawada, K.; Nakamoto, Y.; Kawada, M.; Hida, K.; Matsumoto, T.; Murakami, T.; Hasegawa, S.; Togashi, K.; Sakai, Y. Relationship between 18F-Fluorodeoxyglucose accumulation and KRAS/BRAF mutations in colorectal cancer. Clin. Cancer Res. 2012, 18, 1696–1703, doi:10.1158/1078-0432.ccr-11-1909.
- Linster, C.L.; Schaftingen, E.V. Vitamin C. FEBS J. 2007, 274, 1–22, doi:10.1111/j.1742-4658.2006.05607.x@10.1002.
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nat. Cell Biol. 2006, 441, 437–443, doi:10.1038/nature04871.
- Sinnberg, T.; Noor, S.; Venturelli, S.; Berger, A.; Schuler, P.; Garbe, C.; Busch, C. The ROS-induced cytotoxicity of ascorbate is attenuated by hypoxia and HIF-1alpha in the NCI60 cancer cell lines. J. Cell. Mol. Med. 2014, 18, 530–541, doi:10.1111/jcmm.12207.
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282, doi:10.1038/s41568-019-0135-7.
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nat. Cell Biol. 1983, 301, 89–92, doi:10.1038/301089a0.
- Gama-Sosa, M.A.; Slagel, V.A.; Trewyn, R.W.; Oxenhandler, R.; Kuo, K.C.; Gehrke, C.W.; Ehrlich, M. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983, 11, 6883–6894, doi:10.1093/nar/11.19.6883.
- Li, J.; Huang, Q.; Zeng, F.; Li, W.; He, Z.; Chen, W.; Zhu, W.; Zhang, B. The prognostic value of global DNA hypomethylation in cancer: A meta-analysis. PLoS ONE 2014, 9, e106290, doi:10.1371/journal.pone.0106290.
- Han, L.; Hou, L.; Zhou, M.-J.; Ma, Z.; Lin, D.-L.; Wu, L.; Ge, Y. Aberrant NDRG1 methylation associated with its decreased expression and clinicopathological significance in breast cancer. J. Biomed. Sci. 2013, 20, 52, doi:10.1186/1423-0127-20-52.
- Starczak, M.; Zarakowska, E.; Modrzejewska, M.; Dziaman, T.; Szpila, A.; Linowiecka, K.; Guz, J.; Szpotan, J.; Gawronski, M.; Labejszo, A.; et al. In vivo evidence of ascorbate involvement in the generation of epigenetic DNA modifications in leukocytes from patients with colorectal carcinoma, benign adenoma and inflammatory bowel disease. J. Transl. Med. 2018, 16, 204, doi:10.1186/s12967-018-1581-9.
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nat. Cell Biol. 2013, 500, 222–226, doi:10.1038/nature12362.
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; Van Booven, D.; Harbour, J.W.; Liu, Z.-J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin. Epigenetics 2015, 7, 51, doi:10.1186/s13148-015-0087-z.
- Modrzejewska, M.; Gawronski, M.; Skonieczna, M.; Zarakowska, E.; Starczak, M.; Foksinski, M.; Rzeszowska-Wolny, J.; Gackowski, D.; Olinski, R. Vitamin C enhances substantially formation of 5-hydroxymethyluracil in cellular DNA. Free. Radic. Biol. Med. 2016, 101, 378–383, doi:10.1016/j.freeradbiomed.2016.10.535.
- Peng, D.; Ge, G.; Gong, Y.; Zhan, Y.; He, S.; Guan, B.; Li, Y.; Xu, Z.; Hao, H.; He, Z.-S.; et al. Vitamin C increases 5-hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clin. Epigenetics 2018, 10, 94, doi:10.1186/s13148-018-0527-7.
- Liu, J.; Hong, J.; Han, H.; Park, J.; Kim, D.; Park, H.; Ko, M.; Koh, Y.; Shin, D.-Y.; Yoon, S.-S. Decreased vitamin C uptake mediated by SLC2A3 promotes leukaemia progression and impedes TET2 restoration. Br. J. Cancer 2020, 122, 1445–1452, doi:10.1038/s41416-020-0788-8.
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 function blocks aberrant self-renewal and leukemia progression. Cell 2017, 170, 1079–1095.e20, doi:10.1016/j.cell.2017.07.032.
- Zhao, H.; Zhu, H.; Huang, J.; Zhu, Y.; Hong, M.; Zhu, H.; Zhang, J.; Li, S.; Yang, L.; Lian, Y.; et al. The synergy of Vitamin C with decitabine activates TET2 in leukemic cells and significantly improves overall survival in elderly patients with acute myeloid leukemia. Leuk. Res. 2018, 66, 1–7, doi:10.1016/j.leukres.2017.12.009.
- Pollard, H.B.; A Levine, M.; Eidelman, O.; Pollard, M. Pharmacological ascorbic acid suppresses syngeneic tumor growth and metastases in hormone-refractory prostate cancer. In Vivo 2010, 24, 249–255.
- Polireddy, K.; Dong, R.; Reed, G.; Yu, J.; Chen, P.; Williamson, S.; Violet, P.-C.; Pessetto, Z.; Godwin, A.K.; Fan, F.; et al. High dose parenteral ascorbate inhibited pancreatic cancer growth and metastasis: mechanisms and a phase I/IIa study. Sci. Rep. 2017, 7, 17188, doi:10.1038/s41598-017-17568-8.
- Gan, L.; Camarena, V.; Mustafi, S.; Wang, G. Vitamin C inhibits triple-negative breast cancer metastasis by affecting the expression of YAP1 and synaptopodin 2. Nutrients 2019, 11, 2997, doi:10.3390/nu11122997.
- Zhang, T.; Huang, K.; Zhu, Y.; Wang, T.; Shan, Y.; Long, B.; Li, Y.; Chen, Q.; Wang, P.; Zhao, S.; et al. Vitamin C–dependent lysine demethylase 6 (KDM6)-mediated demethylation promotes a chromatin state that supports the endothelial-to-hematopoietic transition. J. Biol. Chem. 2019, 294, 13657–13670, doi:10.1074/jbc.ra119.009757.
- Wang, T.; Chen, K.; Zeng, X.; Yang, J.; Wu, Y.; Shi, X.; Qin, B.; Zeng, L.; Esteban, M.A.; Pan, G.; et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a Vitamin-C-Dependent manner. Cell Stem Cell 2011, 9, 575–587, doi:10.1016/j.stem.2011.10.005.
- Ebata, K.; Mesh, K.; Liu, S.; Bilenky, M.; Fekete, A.; Acker, M.G.; Hirst, M.; Garcia, B.A.; Ramalho-Santos, M. Vitamin C induces specific demethylation of H3K9me2 in mouse embryonic stem cells via Kdm3a/b. Epigenetics Chromatin 2017, 10, 36, doi:10.1186/s13072-017-0143-3.
- Yong-Hee, R.; Kim, M.; Kim, S.-Y.; Yi, S.-H.; Rhee, Y.-H.; Kim, T.; Lee, E.-H.; Park, C.-H.; Dixit, S.; Harrison, F.E.; et al. Vitamin C Facilitates Dopamine Neuron Differentiation in Fetal Midbrain Through TET1- and JMJD3-Dependent Epigenetic Control Manner. STEM CELLS 2015, 33, 1320–1332, doi:10.1002/stem.1932.
- Hoffer, L.J.; Levine, M.; Assouline, S.; Melnychuk, D.; Padayatty, S.J.; Rosadiuk, K.; Rousseau, C.; Robitaille, L.; Miller, W.H. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann. Oncol. 2008, 19, 1969–1974, doi:10.1093/annonc/mdn377.
- Carr, A.; Vissers, M.C.M.; Cook, J.S. The effect of intravenous vitamin c on cancer- and chemotherapy-related fatigue and quality of life. Front. Oncol. 2014, 4, 283, doi:10.3389/fonc.2014.00283.
- Park, H.; Kang, J.; Choi, J.; Heo, S.; Lee, D.-H. The effect of high dose intravenous Vitamin C during radiotherapy on breast cancer patients’ neutrophil–lymphocyte ratio. J. Altern. Complement. Med. 2020, 26, 1039–1046, doi:10.1089/acm.2020.0138.
- Carr, A.; Cook, J. Intravenous Vitamin C for cancer therapy—Identifying the current gaps in our knowledge. Front. Physiol. 2018, 9, 1182, doi:10.3389/fphys.2018.01182.