Ferroptosis is classified as a non-canonical cell death mechanism. To date, several natural compounds have been discovered to induce ferroptosis in different cancer models.
- non-canonical cell death
- ferroptosis
- natural compounds
1. Ferroptosis
Ferroptosis, firstly discovered by Dixon et al. in 2012 [12], is a non-canonical cell death characterized by an iron-dependent accumulation of lipid reactive oxygen species (ROS), which leads to cell demise [13]. Ferroptosis differs from any other form of regulated cell death. Morphologically, it does not involve any typical apoptotic feature; it is not characterized by cytoplasmatic swelling or disruption of cell membrane, as in necrotic cell death; the formation of typical autophagic vacuoles is not observed [12]. Ferroptotic cells, instead, are morphologically characterized by a distinct shrinkage of mitochondria with enhanced membrane density and decrease/depletion of mitochondrial cristae [12].
Ferroptosis is caused by compounds able to antagonize glutathione peroxidase 4 (GPX4) in a direct way or through the inhibition of Xc− system. Xc− system is an amino acid antiporter responsible for intracellular transport of extracellular cystine by exchanging intracellular glutamate [14] (Figure 1). Once inside the cells, cystine is reduced to cysteine, an essential substrate for glutathione (GSH) synthesis [15]. Hence, the inhibition of Xc− system alters GSH biosynthesis, reducing the antioxidant activity of glutathione and selenium-dependent GPXs [16,17,18]. Among GPXs, GPX4 is the only one able to reduce hydrogen peroxides or organic hydroperoxides into water or corresponding alcohols by converting GSH into oxidized glutathione (GSSG) [19,20] (Figure 1). Then, the inhibition of GPX4, through direct or indirect mechanisms, leads to lipid ROS accumulation and activates the ferroptotic cell death cascade [12,21,22] (Figure 1).
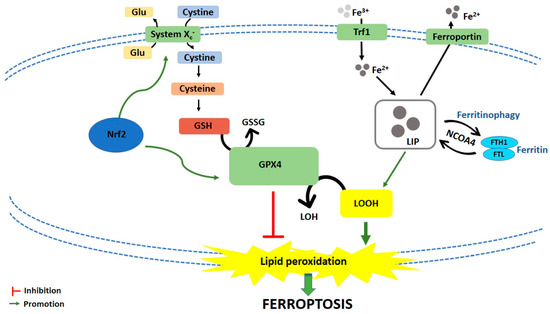
Figure 1. Schematic representation of ferroptotic cell death pathway. Glu: Glutamate; GSH: Glutathione; GSSG: Oxidized glutathione; GPX4: Glutathione peroxidase 4; LOH: Lipid alcohols; LOOH: Lipid hydroperoxides; Nrf2: Nuclear factor (erythroid-derived 2)-like 2; Trf1: Transferrin receptor 1; LIP: Labile iron pool; FTH1: Ferritin heavy chain 1; FTL: Ferritin light chain; NCOA4: Nuclear receptor coactivator.
Iron-dependent accumulation of lipid ROS can occur through non-enzymatic and/or enzymatic lipid peroxidation. Non-enzymatic lipid peroxidation, also called lipid autoxidation, consists in a free radical-driven chain reaction where ROS initiate the oxidation of polyunsaturated fatty acids (PUFAs). Within an autocatalytic process, autoxidation can be propagated leading to membrane destruction, and subsequent ferroptotic cell death [23]. Enzymatic lipid peroxidation is mostly driven by lipoxygenases (LOXs). LOXs, through their dioxygenase activity, catalyze oxygen insertion into PUFAs membrane, generating different lipid hydroperoxides (LOOH), which can start the autocatalytic process of lipid autoxidation mentioned above [22].
If the link between lipid metabolism and ferroptosis induction is well known, how lipid peroxidation leads to ferroptotic cell death is not clear yet. Two mechanisms have been hypothesized. The first hypothesis is that lipid hydroperoxides, produced by PUFAs peroxidation, generate reactive toxic products, i.e., 4-hydroxy-2-nonenal (4-HNE) or malondialdehyde (MDA), which consequently inactivate different survival proteins, leading to ferroptosis [24]. The second hypothesis is that extensive phospholipids peroxidation leads to structural and functional modifications of cellular membrane [23].
2. Natural Compounds as Ferroptosis Inducers
Several natural compounds, alone or in combination, have been found to induce ferroptosis in different in vitro cancer models (Table 1).
Table 1. Natural products as in vitro inducers of ferroptosis.
| Compound | Compound Source | Cell Line(s) | Concentrations (Where Specified) |
Time (Where Specified) | Ferroptosis Markers | Supplementary Effects |
|---|---|---|---|---|---|---|
| Actinia chinensis (Planch), drug-containing rat serum | Actinia chinensis Planch | HGC-27 | 90, 180 and 360 mg/mL | 48 h | ↓ Cell proliferation | |
| 24 and 48 h | ↓ Cell migration | |||||
| 180 mg/mL | 48 h | ↑ ROS | ↓ after Ferr-1 treatment | |||
| 90, 180 and 360 mg/mL | ↓ GPX4 | |||||
| ↓ xCT | ||||||
| Albiziabioside A | Albizia inundata Mart. | MCF-7 | 10 μM | 24 h | ↑ Cytotoxicity | ↑ after Fe2+ treatment |
| ↓ after Ferr-1 treatment | ||||||
| ↓ after DFO treatment | ||||||
| ↓ after vitamin E treatment | ||||||
| / | ↑ ROS | |||||
| 24 h | ↓ GSH/GSSG ratio | |||||
| 48 h | ↓ GPX4 protein expression | |||||
| / | ↑ MDA | |||||
| ↑ Lipid peroxides | ||||||
| Amentoflavone | Selaginella spp. and other plants | U251, U373 | 10 and 20 μM | / | ↑ Fe2+ | |
| ↓ FTH | ↑ after ATG7 knockdown | |||||
| ↑ MDA | ↓ after FTH overexpression | |||||
| ↑ Lipid ROS | ↓ after FTH overexpression | |||||
| ↓ after BafA1 treatment | ||||||
| ↓ after ATG7 knockdown | ||||||
| ↓ GSH | ↓ after FTH overexpression | |||||
| ↓ after BafA1 treatment | ||||||
| ↓ after ATG7 knockdown | ||||||
| 20 μM | ↑ Cell death ratio (%) | ↓ after Ferr-1 treatment | ||||
| ↓ after DFO treatment | ||||||
| ↓ after FTH overexpression | ||||||
| ↓ after BafA1 treatment | ||||||
| ↓ after ATG7 knockdown | ||||||
| Ardisiacrispin B | Ardisia kivuensis Taton | CCRF-CEM | 0.59, 0.93, 2.33, 4.66, 9.32, 18.64 and 37.28 μM | 24 h | ↑ Cytotoxicity | ↓ after Ferr-1 treatment |
| ↓ after DFO treatment | ||||||
| 0.3, 0.6, 1.2 and 2.4 μM | ↑ ROS | |||||
| Aridanin | Tetrapleura tetraptera (Schum. & Thonn) Taub. | CCRF-CEM | 1, 2, 4, 8, 15, 30 and 61 μM | 24 h | ↓ Cell viability | ↓ after Ferr-1 treatment |
| ↓ after DFO treatment | ||||||
| Artenimol (artemisinin semi-syntethic derivative) |
Artemisia annua L. | CCRF-CEM | 0.01, 0.1, 1, 10 and 100 μM | / | ↓ Cell viability | ↓ after Ferr-1 treatment |
| ↓ after DFO treatment | ||||||
| Artesunate (artemisin semi-synthetic derivative) | Artemisia annua L. | DAUDI, CA-46 | 4 and 20 μM | 48 h | ↓ Cell viability | ↑ after DFO treatment |
| ↑ after Ferr-1 treatment | ||||||
| ↑ after Lip-1 treatment | ||||||
| ↑ after down-regulation of CHAC1 expression | ||||||
| 5, 10 and 20 μM | 24 and 48 h | ↑ ROS | ||||
| ↑ Lipid peroxidation | ↓ after down-regulation of CHAC-1 expression | |||||
| 5, 10 and 20 μM | 24 h | ↑ CHAC1, ↑ ATF4, ↑ CHOP protein expression | ||||
| MT-2 | 50 μM | 24 h | ↑ ROS | |||
| 0.4, 2 and 10 μM | ↑ Cytotoxicity | ↓ after DFO treatment | ||||
| 2 and 10 μM | ↓ after Ferr-1 treatment | |||||
| HUT-102 | 50 μM | 24 h | ↑ ROS | ↓ after NAC treatment | ||
| 2 and 10 μM | ↑ Cytotoxicity | ↓ after DFO treatment | ||||
| 10 and 50 μM | ↓ after Ferr-1 treatment | |||||
| HN9 | 50 μM | 72 h | ↓ Cell viability | ↓ after HTF treatment | ||
| ↑ after DFO treatment | ||||||
| ↑ after Trolox treatment | ||||||
| 2.5 and 5 μM | ↑ after Keap1 knockdown | |||||
| ↑ after Nrf2 knockdown | ||||||
| 50 μM | 24 h | ↑ ROS | ↓ after Ferr-1 treatment | |||
| ↓ after Trolox treatment | ||||||
| ↑ Lipid ROS | ↓ after Ferr-1 treatment | |||||
| ↓ after Trolox treatment | ||||||
| HN9, HN9-cisR | 10, 25 and 50 μM | 24 h | ↑ Nrf2 protein expression | |||
| ↓ xCT, ↓ RAD51, ↓ Keap1 protein expression | ||||||
| HN9-cisR, HN3-cisR, HN4-cisR | 10, 25 and 50 μM | 24 h | ↑ Nrf2, ↑ HO-1, ↑ NQO1 protein expression | |||
| ↓ Keap1 protein expression | ||||||
| 50 μM | ↑ Nrf2, ↑ HO-1, ↑ NQO1 mRNA levels | |||||
| HN3-cisR | 25 and 50 μM | 24 h | ↓ GSH | ↓ after trigonellin treatment | ||
| ↑ after Trolox treatment | ||||||
| ↑ after Nrf2 knockdown | ||||||
| ↑ ROS | ↓ after Trolox treatment | |||||
| ↑ after Nrf2 knockdown | ||||||
| ↑ Lipid ROS | ↓ after trigonellin treatment | |||||
| ↓ Cell viability | ↓ after Nrf2 knockdown | |||||
| ↓ after HO-1 knockdown | ||||||
| ↑ after Trolox treatment | ||||||
| PaTU8988, AsPC-1 | 20 μM | 24 h | ↓ Cell viability | ↑ after Ferr-1 treatment | ||
| ↑ after GRP78 overexpression | ||||||
| ↓ after GRP78 knockdown | ||||||
| ↑ MDA | ↓ after DFO treatment | |||||
| ↓ after Ferr-1 treatment | ||||||
| ↓ after GRP78 overexpression | ||||||
| ↑ after GRP78 knockdown | ||||||
| ↑ Lipid peroxidation | ↓ after Ferr-1 treatment | |||||
| 10, 20 and 40 μM | ↑ GRP78 mRNA levels | |||||
| ↑ GRP78 protein expression | ||||||
| HEY1 | 25 and 50 μM | 48 h | ↑ Cell death | ↓ after Ferr-1 treatment | ||
| ↓ after DFO treatment | ||||||
| ↑ after HT treatment | ||||||
| HEY2 | 100 μM | ↑ Cell death | ↓ after Ferr-1 treatment | |||
| HEY2, SKOV3 | 50 and 100 μM | ↑ Cell death | ↓ after DFO treatment | |||
| ↑ after HT treatment | ||||||
| HEY1, HEY2, SKOV-3 | 10, 25, 50 and 100 μM | 24 h | ↑ ROS | ↓ after GSH treatment | ||
| HEY1, HEY2, SKOV-3, OVCAR8, TOV-112D, TOV-21G | 25, 50 and 100 μM | 48 h | ↑ Cell death | ↓ after GSH treatment | ||
| Panc-1 | 50 μM | 24 h | ↑ ROS | ↓ after Trolox treatment | ||
| ↓ Colony formation | ↑ after DFO treatment | |||||
| ↑ after Trolox treatment | ||||||
| ↑ after Ferr-1 treatment | ||||||
| ↓ after HTF treatment | ||||||
| ↑ HO-1 protein expression | ||||||
| ↑ Lipid peroxidation | ↓ after Trolox treatment | |||||
| ↓ after Ferr-1 treatment | ||||||
| Panc-1, COLO-357 | 48 h | ↑ Cell death | ↓ after Ferr-1 treatment | |||
| BxPC-3, Panc-1 | 24 and 48 h | ↑ Cell death | ↓ after DFO treatment | |||
| BxPC-3, Panc-1, AsPC-1 | ↑ Cell death | ↑ after HTF treatment | ||||
| Betula etnensis Raf. methanolic extract | Betula etnensis Raf. | CaCo2 | 5, 50, 250 and 500 μg/mL | 72 h | ↓ Cell viability | |
| 5, 50 and 250 μg/mL | ↑ LDH release | |||||
| ↑ ROS | ||||||
| ↑ LOOH | ||||||
| ↓ RSH | ||||||
| 5 and 50 μg/mL | ↓ HO-1 levels | |||||
| 250 μg/mL | ↑ HO-1 levels | |||||
| D13 (albiziabioside A derivative) | Albizia inundata Mart. | HCT116 | 0.31, 1.25 and 5 μM | / | ↑ Cytotoxicity | ↓ after Ferr-1 treatment |
| ↓ after DFO treatment | ||||||
| ↑ after Fe2+ treatment | ||||||
| ↑ after Fe3+ treatment | ||||||
| 48 h | ↓ GPX4 protein expression | |||||
| / | ↑ MDA | |||||
| Dihydroartemisinin (artemisin semi-synthetic derivative) | Artemisia annua L. | THP-1 | 5, 10 and 15 μM | 12 h | ↓ Cell viability | |
| ↑ ROS | ||||||
| HL-60 | 5, 10 and 15 μM | 12 h | ↓ Cell viability | ↑ after Ferr-1 treatment | ||
| ↑ after DFO treatment | ||||||
| ↑ after NAC treatment | ||||||
| ↑ after BafA1 treatment | ||||||
| ↑ after 3-MA treatment | ||||||
| ↑ after ATG7 knockdown | ||||||
| ↑ after FTH overexpression | ||||||
| ↑ after ISCU overexpression | ||||||
| ↑ Lipid ROS | ↓ after ATG7 knockdown | |||||
| ↓ after FTH overexpression | ||||||
| ↓ GSH | ↑ after ISCU overexpression | |||||
| ↑ ROS | ↓ after DFO treatment | |||||
| ↓ after NAC treatment | ||||||
| ↓ after ISCU overexpression | ||||||
| ↑ IRP2 protein expression | ||||||
| ↓ FTH, ↓ GPX4 protein expression | ↑ after ISCU overexpression | |||||
| ↑ after BafA1 treatment | ||||||
| G0101, G0107 | 10, 20, 40, 80 and 160 μM | 24 h | ↑ ROS | |||
| 20, 40, 80 and 160 μM | ↑ Lipid ROS | |||||
| ↑ MDA | ||||||
| ↓ GSH | ||||||
| ↑ GSSG | ||||||
| ↑ Cell death | ↓ after DFO treatment | |||||
| ↓ after Ferr-1 treatment | ||||||
| ↓ after Lip-1 treatment | ||||||
| U251 U373 |
5, 10, 20 and 40 μM 20, 40, 80 and 160 μM |
24 h | ↓ GSH | |||
| U251 U373 |
2.5, 5, 10, 20 and 40 μM 10, 20, 40, 80 and 160 μM |
24 and 48 h | ↑ ROS | ↓ after DFO treatment | ||
| ↑ after PERKi treatment | ||||||
| ↑ after ATF4 siRNA treatment | ||||||
| ↑ after HSPA5 siRNA treatment | ||||||
| U251 U373 |
2.5, 5, 10 and 20 μM 10, 20, 40, 80 and 160 μM |
3, 6, 12, 24 and 48 h | ↑ Lipid ROS | ↑ after ATF4 siRNA treatment | ||
| ↑ after HSPA5 siRNA treatment | ||||||
| U251 U373 |
5, 10, 20 and 40 μM 80 μM |
3, 6, 12, 24 and 48 h | ↑ MDA | ↑ after PERKi treatment | ||
| ↑ after ATF4 siRNA treatment | ||||||
| ↑ after HSPA5 siRNA treatment | ||||||
| U251 U373 |
10, 20 and 40 μM 40, 80 and 160 μM |
48 h | ↑ Cell death | ↓ after DFO treatment | ||
| ↓ after Ferr-1 treatment | ||||||
| ↓ after Lip-1 treatment | ||||||
| ↑ after PERKi treatment | ||||||
| ↑ after ATF4 siRNA treatment | ||||||
| ↑ after HSA5 siRNA treatment | ||||||
| Dihydroisotanshinone I | Salvia miltiorrhiza Bunge | MCF-7 | 5 and 10 μM | 24 h | ↓ GPX4 activity | |
| MCF-7, MDA-MB231 | 10 μM | ↓ GPX4 protein expression | ||||
| 5 and 10 μM | ↑ MDA | |||||
| 10 μM | ↓ GSH/GSSG ratio | |||||
| Epunctanone | Garcinia epunctata Stapf. | CCRF-CEM | 1.04, 1.66, 4.14, 8.28, 16.56, 33.11 and 66.23 μM | 24 h | ↑ Cytotoxicity | ↓ after Ferr-1 treatment |
| ↓ after DFO treatment | ||||||
| 2.95, 5.91, 11.81 and 23.63 μM | ↑ ROS | |||||
| Erianin | Dendrobium chrysotoxum Lindl | H460, H1299 | 50 and 100 nM | 24 h | ↑ Cell death | ↓ after NAC treatment |
| ↓ after Ferr-1 treatment | ||||||
| ↓ after Lip-1 treatment | ||||||
| ↓ after GSH treatment | ||||||
| 50 and 100 nM | / | ↑ ROS | ||||
| ↓ GSH | ||||||
| 12.5, 25, 50 and 100 nM | ↑ MDA | |||||
| ↑ HO-1, ↑ transferrin protein expression | ||||||
| ↓ GPX4, ↓ CHAC2, ↓ SLC40A1, ↓ SLC7A11 protein expression | ||||||
| 5, 10 and 25 μM | 24 h | ↑ Ca2+ levels | ||||
| ↑ Calmodulin protein expression | ||||||
| Ferroptocide (pleuromutilin semi-syntetic derivative) | Pleurotus passeckerianus; Drosophila subatrata; Clitopilus scyphoides, and others spp. | ES-2 | 5, 10 and 25 μM | 1 h | ↑ ROS | ↓ after DFO treatment |
| 10 and 25 μM | ↑ Mitochondrial ROS | |||||
| 10 μM | ↑ Lipid ROS | ↓ after DFO treatment | ||||
| 5, 10 and 25 μM | 14 h | ↑ Cell death | ↓ after Ferr-1 treatment | |||
| ↓ after DFO treatment | ||||||
| ↓ after Trolox treatment | ||||||
| HCT116 | 5, 10 and 25 μM | 10, 24 and 48 h | ↑ Cell death | ↓ after DFO treatment | ||
| ↓ after Trolox treatment | ||||||
| ↓ after NAC-1 treatment | ||||||
| ↑ after TXN knockdown | ||||||
| ↓ after Ferr-1 treatment | ||||||
| 5, 10 and 25 μM | 1.5 and 72 h | ↑ ROS | ↑ after TXN knockdown | |||
| 10 μM | 2 and 72 h | ↑ Lipid ROS | ↑ after TXN knockdown | |||
| ↓ after DFO treatment | ||||||
| 4T1 | 5, 10 and 25 μM | 18 h | ↑ Cell death | ↓ after DFO treatment | ||
| ↓ after Ferr-1 treatment | ||||||
| 10 μM | 2 h | ↑ Lipid ROS | ↓ after Ferr-1 treatment | |||
| ↓ after DFO treatment | ||||||
| HT-29 | 5, 10 and 25 μM | 12 h | ↑ Cell death | ↓ after DFO treatment | ||
| ↓ after Trolox treatment | ||||||
| ↓ after NAC treatment | ||||||
| ↓ after Ferr-1 treatment | ||||||
| Gallic Acid | Natural polyhydroxy phenolic compound, found in various foods | HeLa | 50 μg/mL | 12 h | ↑ Lipid peroxidation | |
| HeLa, H446, SHSY-5Y | 36 h | ↑ Cell death | ↓ after DFO treatment | |||
| A375, MDA-MB-231 | 10, 25, 50, 100 and 200 μg/mL | 24 h | ↓ Cell viability | |||
| MDA-MB-231 | 25 μg/mL | ↑ ROS | ||||
| A375 | 50 μg/mL | |||||
| MDA-MB-231 | / | / | ↓ GPX4 activity | |||
| A375, MDA-MB-231 | / | / | ↑ MDA | |||
| Physcion 8-O-β-glucopyranoside | Rumex japonicus Houtt. | MGC-803, MKN-45 | 10, 20, 30, 40 and 50 μM | 24, 48, 72 and 96 h | ↓ Cell viability | ↑ after Ferr-1 treatment |
| ↑ after GPNA treatment | ||||||
| ↑ after 968 treatment | ||||||
| ↓ after GLS2 knockdown | ||||||
| / | / | ↓ Cell proliferation | ↑ after miR-103a-3p overexpression | |||
| / | 24 h | ↓ Cell invasion | ↑ after miR-103a-3p overexpression | |||
| ↓ Cell migration | ↑ after miR-103a-3p overexpression | |||||
| / | / | ↑ Lipid ROS | ↓ after GPNA treatment | |||
| ↓ after 968 treatment | ||||||
| ↓ after GLS2 knockdown | ||||||
| ↓ after miR-103a-3p overexpression | ||||||
| ↑ MDA | ↓ after GPNA treatment | |||||
| ↓ after 968 treatment | ||||||
| ↓ after GLS2 knockdown | ||||||
| ↓ after miR-103a-3p overexpression | ||||||
| ↑ Fe2+ | ↓ after GPNA treatment | |||||
| ↓ after 968 treatment | ||||||
| ↓ after GLS2 knockdown | ||||||
| ↓ after miR-103a-3p overexpression | ||||||
| ↓ miR-103a-3p expression | ||||||
| ↑ GLS2 protein levels | ↓ after miR-103-3p transfection | |||||
| Piperlongumine | Piper Longum L. | Panc-1 | 4, 6, 8, 10, 12 and 14 μM | 16 h | ↓ Cell viability | ↑ after NAC treatment |
| ↑ after Ferr-1 treatment | ||||||
| ↑ after Lip-1 treatment | ||||||
| ↑ after DFO treatment | ||||||
| MIAPaCa-2 | 10 μM | 16 h | ↓ Cell viability | ↑ after CPX treatment | ||
| ↑ after PD146176 treatment | ||||||
| 4 h | ↓ GSH | |||||
| Progenin III | Raphia vinifera P. Beauv | CCRF-CEM | 2, 3, 7, 14 and 55 μM | 24 h | ↓ Cell viability | ↑ after Ferr-1 treatment |
| ↑ after DFO treatment | ||||||
| 1.59 and 3.18 μM | ↑ ROS | |||||
| Ruscogenin | Ruscus aculeatus L. Radix Ophiopogon japonicas (Thunb.) Ker Gawl. | BxPC-3, SW1990 | 7 μM | / | ↑ Cell death | ↑ after FAC treatment |
| ↓ after DFO treatment | ||||||
| / | 6 h | ↑ Cell death | ↓ after transferrin knockdown | |||
| ↓ after ferroportin overexpression | ||||||
| 3 and 7 μM | 12 and 24 h | ↑ Fe2+ | ↓ after DFO treatment | |||
| 1, 2, 4, 6 and 24 h | ↑ ROS | ↓ after DFO treatment | ||||
| 6 and 12 μM | 24 h | ↑ Transferrin | ||||
| ↓ Ferroportin | ||||||
| Solasonine | Solanum melongena L. | HepG2 | 15 ng/mL | 24 h | ↑ Cell death | ↓ after Ferr-1 treatment |
| ↓ after DFO treatment | ||||||
| ↑ Lipid ROS | ↓ after Ferr-1 treatment | |||||
| ↓ after DFO treatment | ||||||
| ↓ GSS, ↓ GPX4 mRNA levels | ||||||
| ↓ GSS, ↓ GPX4 protein expression | ||||||
| Typhaneoside | Pollen Typhae | Kas-1, HL-60, NB4 | 40 μM | 24 h | ↓ Cell viability | ↑ after Ferr-1 treatment |
| ↑ after DFO treatment | ||||||
| ↑ after 3-MA treatment | ||||||
| ↑ after BafA1 treatment | ||||||
| ↑ after Z-VAD-FMK treatment | ||||||
| ↑ after rapamycin treatment | ||||||
| ↑ after ATG7 knockdown | ||||||
| 20, 30 and 40 μM | ↑ ROS | ↓ after DFO treatment | ||||
| ↓ after NAC treatment | ||||||
| ↓ GSH | ||||||
| ↑ Lipid ROS | ↓ after ATG7 knockdown | |||||
| ↓ after BafA1 treatment | ||||||
| ↓ GPX4, ↓ FTH mRNA levels | ||||||
| ↑ IRP2 mRNA levels | ||||||
| Ungeremine | Crinum zeylanicum L. | CCRF-CEM | 2.37, 3.76, 9.40, 18.79, 37.58, 75.17 and 150.33 μM | 24 h | ↓ Cell proliferation | ↑ after Ferr-1 treatment |
| ↑ after DFO treatment | ||||||
| 1.22, 2.45, 4.89 and 9.78 μM | ↑ ROS | |||||
| Whitaferin A | Withania somnifera (L.) Dunal | IMR-32 | / | / | ↑ ROS | |
| 1 and 10 μM | 2, 4, 8, 12 and 24 h | ↓ GPX4 expression | ||||
| 10 μM | 3 and 5 h | ↓ GPX4 activity | ||||
| / | / | ↑ Lipid peroxidation | ↓ after DFO treatment | |||
| 1 μM | 4, 8 and 12 h | ↑ Fe2+ | ↑ after hemin treatment | |||
| 1, 2, 4, 8, 12 and 24 h | ↑ HO-1, ↑ Keap1, ↑ Nrf2 protein expression | |||||
| 6, 8, 12 and 16 h | ↑ Cell death | ↓ after GPX4 overexpression | ||||
| ↓ after ZnPP treatment | ||||||
| ↓ HO-1 knockdown | ||||||
| ↑ after hemin treatment | ||||||
| IMR-32, SK-N-SH | 1 and 10 μM | 6, 8, 12 and 16 h | ↑ Cell death | ↓ after Ferr-1 treatment | ||
| ↓ after CPX treatment | ||||||
| ↓ after α-tocopherol treatment | ||||||
| ↓ after UOI26 treatment | ||||||
| ↓ after Flt3 inhibitor treatment | ||||||
| 1 μM | / | Nrf2 pathway activation | ||||
| / | ↑ FTH1, ↑ HO-1 gene expression | |||||
| 1, 2, 4, 8, 12 and 24 h | ↑ FTH1, ↑ HO-1 mRNA levels | |||||
| WA-NPs | Withania somnifera L. Dunal | IMR-32 | 1 and 10 μM | 8, 10, 12, 16, 20 and 24 h | ↑ Cell death |
Abbreviations: ↑: Increase; ↓: Decrease; 3-MA:3-methyladenine; 968: Compound 968, GLS2 inhibitor;; ATF4: Activating transcription factor 4; ATG7: Autophagy related 7; BafA1: Bafilomycin 1; CHAC1: Glutathione-specific Gamma-glutamylcyclotransferase 1; CHOP: CCAAT/enhancer-binding protein homologous protein; CPX: Ciclopirox, intracellular iron chelator; DFO: Deferoxamine; FAC: Ferric ammonium citrate; Fe2+: Ferrous ion; Fe3+: Ferric ion; Ferr-1: Ferrostatin-1; Flt3: Receptor tyrosine kinase fms-like tyrosine kinase 3; FTH: Ferritin heavy chain; FTH1: Ferritin heavy chain 1; GLS2: Glutaminase 2; GPNA: Glutamine transporter inhibitor; GPX4: Glutathione peroxidase IV; GSH: Glutathione; GSS: Glutathione synthetase; GSSG: Oxidized glutathione; HO-1: Heme oxygenase 1; HN3-cisR: Cisplatin-resistant HN3 cells; HN4-cisR: Cisplatin-resistant HN4 cells; HN9-cisR: Cisplatin-resistant HN9 cells; HSPA5: Heat shock protein family A (Hsp70) member 5; HTF: Holo-transferrin; IRP2: Iron regulator protein 2; ISCU: Iron-sulfur cluster assembly enzyme; Keap1: Kelch-like ECH-associated protein 1; Lip-1: Liproxstatin-1; MDA: Malondialdehyde; NAC: N-acetylcysteine; NQO1: NAD(P)H quinone dehydrogenase 1; Nrf2: Nuclear factor erythroid 2–related factor 2; PD146176: Lypoxygenase inhibitor; PERKi: PERK inhibitor I (GSK2606414); ROS: Reactive oxygen species; RSH: Thiols; Spp.: Species; TXN: Thioredoxin; WA-NPs: Whitaferin A nanoparticles; xCT: Cystine/glutamate antiporter; ZnPP: Zinc protoporphyrin, HO-1 inhibitor; Z-VAD-FMK: Pan-caspase inhibitor.
Amentoflavone is a flavonoid mainly found in Selaginella tamariscina (P. Beauv.) Spring and in other species of Selaginella, as well as in many other plant species [55]. Amentoflavone exhibits anticancer effects in several tumor cells by inducing apoptosis, autophagy and ferroptosis, and by inhibiting cell-cycle progression [27,56,57,58,59,60,61]. In U251 and U373 glioma cell lines and in a glioma xenograft model, but not in normal human astrocytes, it triggered ferroptotic cell death by reducing GSH and ferritin heavy chain (FTH) intracellular levels, thus leading to the accumulation of lipid ROS and malondialdehyde (MDA), a PUFAs oxidation product, and subsequent cell death [27] (Table 1). Hence, amentoflavone induces ferroptosis through the rupture of iron homeostasis by reducing the intracellular levels of FTH, which is involved in the intracellular iron storage [27]. Interestingly, both in vitro and in vivo, amentoflavone induced the degradation of FTH by activating autophagy via AMPK (AMP-activated protein kinase)/mTOR (mammalian target of rapamycin)/P70S6K (phosphoprotein 70 ribosomial protein S6 kinase) signaling pathway, suggesting the induction of autophagy-dependent ferroptosis [27]. Autophagy is known as a potent ferroptosis enhancer. Ferritinophagy, in particular, degrades the iron storage protein ferritin and increases the release of free iron, leading to ferroptosis induction [62,63,64].
Two other natural compounds that trigger autophagy-dependent ferroptosis are dihydroartemisinin (DHA) and typhaneoside. DHA is a semi-synthetic derivative of artemisinin, a sesquiterpene lactone derived from Artemisia annua L. currently used as antimalarial agent, which promotes ferroptosis in glioma cells [40] and ferroptosis together with apoptosis in acute myeloid leukemia (AML) cancer cells [39] (Table 1). Typhaneoside, a flavonoid found in the extract of Pollen Typhae, triggered apoptotic and ferroptotic cell death in AML cancer cells [52] (Table 1). In particular, in AML cancer cells, as for amentoflavone, both DHA and typhaneoside induced autophagy-dependent ferroptosis [39,52] by raising the degradation of ferritin through ferritinophagy; moreover, autophagy inhibition mitigated ferroptosis induction by the two natural compounds [39,52] (Table 1). In another experimental setting, DHA did not trigger ferroptosis itself, but it sensitized resistant cancer cells to ferroptosis. In particular, in vitro [mouse embryonic fibroblasts (MEFs) and human osteosarcoma HT1080 cells] and in vivo (GPX4 iKO H292-xenografted female athymic nude-Foxn 1nu/Foxn1+ mice), DHA perturbed iron homeostasis leading to an increase in intracellular iron levels, which concurred to the restoration of RSL3′s and erastin’s ability to induce ferroptosis (Table 1) [65].
Artesunate is another semi-synthetic derivative of artemisinin. It induces ferroptosis in pancreatic [34,36], ovarian [35], head and neck cancer (HNC) [33], T-cell leukemia/lymphoma (ATLL) [32], and in Burkitt’s lymphoma [31] through the modulation of different molecular targets (Table 1). One of these targets is the endoplasmic reticulum (ER). ER stress is a condition of oxidative stress and perturbations in the ER folding machinery provoked by the accumulation of unfolded/misfolded proteins. ER stress activates a signaling process, called unfolded protein response (UPR), in order to lessen ER stress and to restore ER homeostasis [66,67]. In DAUDI and CA-46 lymphoma cells, artesunate triggered ferroptosis and ER stress through the activation of ATF4 (activating transcription factor-4)-CHOP (C/EBP [CCAAT-enhancer-binding protein] homologous protein)-CHAC1 (glutathione-specific γ-glutamylcyclotransferase 1) pathway and PERK [protein kinase RNA (PKR)-like ER kinase] branch of UPR [31] (Table 1). As proposed by the authors [31], the upregulation of CHAC1 possessing a GSH degradation activity [68,69] probably contributes to artesunate-induced ferroptosis [31]. Besides, it is well known that ATF4 could be upregulated by the depletion of amino acids [70], such as that of intracellular cysteine caused by ferroptosis inducers through the system Xc− inhibition. Hence, artesunate might induce ER stress in Burkitt’s lymphoma cells by altering the system Xc−, even if it has to be confirmed. ER stress is also involved in artesunate-induced ferroptosis in KRas mutant pancreatic cancer cells (PaTU8988 and AsPC-1) and in AsPC-1 xenografted BALB/c nude mice [34] (Table 1 and Table S2). Indeed, knockdown of glucose-regulated protein 78 (GRP78), which is considered the master regulator of the UPR signaling process [71], and the inhibition of the three UPR transducers [PERK, IRE1 (inositol requiring protein-1) and ATF6 (activating transcription factor-6] [72], enhanced artesunate-induced ferroptosis in vitro and in vivo [34] (Table 1). Of note, artesunate triggered ferroptosis in a most efficient way in pancreatic cancer cells carrying mutationally-active KRas mutations (i.e., AsPC-1) rather than in pancreatic cancer cells expressing wild type KRas (i.e., COLO-357 and BxPC-3) [36]. This outcome is not odd since KRas mutation often leads to low antioxidant ferritin and transferrin levels and increased number of transferrin receptors and may sensitize pancreatic adenocarcinomas to ferroptosis [33,73]. Still, given that KRas mutant tumors are hardly druggable, these results are quite auspicious [33,74,75].
The role of nuclear factor (erythroid-derived 2)-like 2 (Nrf-2) in ferroptosis is still a matter of debate. Normally, Nrf2 is kept inactivated by Kelch-like ECH-associated protein 1 (Keap-1). Under increased oxidative stress conditions, Nrf2 dissociates from Keap1, translocates into the nucleus, and starts the transcription of the so-called antioxidant responsive element (ARE)-dependent genes [76,77,78]. Most of the Nrf2 target genes are involved in the maintenance of redox homeostasis [79,80], including the regulation of system Xc− [81,82,83], and also in iron and heme homeostasis. They regulate heme-oxygenase 1 (HO-1), ferroportin, and light chain and heavy chain of ferritin (FTL/FTH1) [76,84,85]. In other words, Nrf2 activation could be considered a negative ferroptosis regulator since it endorses antioxidant elements and iron storage, and limits cellular ROS production [86]. Furthermore, since it has been shown that ferroptosis inducers capable of activating Nrf2 pathway promote cellular adaptation and survival and render cancer cells less sensitive to ferroptosis induction themselves [87,88,89], it could be thought that they cannot be considered good candidates for anticancer therapy. However, the activation of Nrf2 pathway could promote ferroptotic cell death. Shifting the focus from the antioxidant properties of Nrf2 effectors to their ability in increasing intracellular iron content, that evidence is not surprising. For instance, HO-1 is responsible for heme catabolism, which produces iron, monoxide, and biliverdin. Thus, it is plausible assuming that the Nrf2 antioxidant response cannot balance the strong iron production, which leads cells to ferroptosis [90]. Accordingly, Kwon et al. [90] demonstrated that hemin, the most prevalent heme metabolite originated by HO-1 catabolism, induced lipid peroxidation as a consequence of iron increase [90]. The opposite role of Nrf2 in ferroptosis seems to be cell-type specific [91], since the activation of Nrf2 pathway protected hepatocellular carcinoma cells against ferroptosis [87], while it promoted ferroptosis in neuroblastoma [54]. Taken together, those results support the hypothesis that Nrf2 could act as a double-edge sword. Even if further studies are needed to disentangle this knot, artesunate supports this hypothesis inducing different effects in different cell lines.
In HNC cells, but not in human oral keratinocytes and fibroblasts, artesunate decreased GSH intracellular levels and increased lipid ROS production and led to ferroptosis [33] (Table 1). However, in HNC cells and cisplatin-resistant HNC cells, artesunate activated the Nrf2 pathway [33] (Table 1), favoring the onset of ferroptosis resistance. As a matter of fact, Keap1 silencing decreased cancer cells’ sensitivity towards artesunate-mediated ferroptosis in both resistant and non-resistant cells, while Nrf2 silencing restored the ability of inducing ferroptosis [33]. In Panc-1 pancreatic cancer cells, induction of ferroptosis by artesunate was accompanied by an increase of HO-1 protein expression [36] (Table 1), which authors associated with the ability of artesunate to increase ROS levels, that in turn activates Nrf2-mediated antioxidant response. Hence, it could be postulated that artesunate induces ferroptosis in pancreatic cancer cells through the HO-1-mediated enhancement of intracellular labile iron (LIP) (i.e., ionic Fe complexes that are redox active). Promotion of ferroptosis by artesunate has been reported also in vivo. In Burkitt’s lymphoma xenograft model, it suppressed tumor growth by inducing lipid peroxidation [31].
Withaferin A (WA) is a naturally occurring steroidal lactone derived from Withania somnifera, a medicinal plant used in Ayurvedic medicine [92]. In a variety of cancer cells, WA showed to exhibit anticancer activity through a plethora of mechanisms, including proteasome and cell-cycle inhibition, modulation of oxidative stress, and induction of apoptosis [92]. In neuroblastoma cells, WA promoted ferroptosis through a dual mechanism: at high dose (10 μM), WA directly binds and inactivates GPX4, thus inducing canonical ferroptosis; at lower dose (1 μM), WA targets Keap1 and activates the Nrf2 pathway, leading to an excessive upregulation of HO-1 and a subsequent LIP increase [54] (Table 1). Through these two mechanisms, WA also promoted ferroptosis and eradicated neuroblastoma xenografts in BALB/c mice [54]. Of note, WA outperformed the full-blown chemotherapeutic agent etoposide both in vitro and in vivo. In vitro, WA efficiently killed a panel of high-risk and etoposide-resistant neuroblastoma cells by inducing ferroptosis [54] (Table 1). In vivo, WA intratumoral administration showed the same efficacy of etoposide in suppressing tumor growth [54]. Most importantly, in contrast to etoposide, WA treatment also repressed neuroblastoma relapse rates in four out of five mice [54]. Hence, taking into account that WA-induced cell death was associated with CD45-positive immune cells infiltration in tumor tissue [54], we could speculate that WA could have activated the immune system, thus inducing an anticancer vaccine effect. This result may itself be a further step in demonstrating that ferroptosis possesses an immunogenic nature, especially in light of the recent confirmation that ferroptosis could promote antitumor immunity [93]. The only sore point of the study, together with the small number of animals used in the experimentation, was the toxic effect of WA observed in vivo. Since upon WA-systemic injection severe weight loss–related adverse effects were detected and given the scarce water solubility of WA, authors formulated WA-encapsulated nanoparticles (WA-NPs) [54]. WA-NPs showed the same efficacy of non-encapsulated WA in vitro (Table 1) and in vivo, constraining systemic side-effects induced by WA, thus allowing systemic application and an effective tumor targeting of WA [54].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13020304