Immunotherapy is now the preferred treatment for most lung cancer patients. It is used to treat unresectable stage III non-small-cell lung cancer and is the first-line therapy for non-oncogene-driven advanced/metastatic non-small-cell lung cancer patients (either alone or in combination with chemotherapy). The binding of PD-1 to its ligands (PD-L1 or PD-L2) on tumor cells suppresses T cells through a negative feedback loop, thereby leading to evasion of the immune response. Unfortunately, most patients that respond initially to immunotherapy develop resistance over time, thus limiting the durability of immunotherapy.
- lung cancer
- PD-1
- PD-L1
- immunotherapy
- immune checkpoint inhibitors
- acquired resistance
1. Introduction
Immunotherapy represents a new paradigm for the treatment of non-small-cell lung cancers (NSCLC), with immune checkpoint inhibitors (ICI) such as anti-programmed death-1 (PD-1)/PD-ligand-1 (PD-L1) leading the field. The binding of PD-1 to its ligands (PD-L1 or PD-L2) on tumor cells suppresses T cells through a negative feedback loop, thereby leading to evasion of the immune response [1][2]. Although antibodies targeting PD-1 or PD-L1 have demonstrated impressive and durable clinical efficacy in a subset of patients with NSCLC, many patients do not respond at all (primary resistance), whilst others that initially respond ultimately relapse (acquired resistance) (Figure 1) [3].
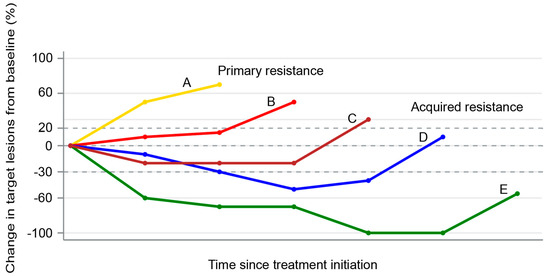
Figure 1. Spider plot showing examples of primary (patient A) and acquired resistance (patients B–D) to immune checkpoint inhibitors. Patient A represents disease progression as the best response. Patients B and C represent initial stable disease followed by progression. Patient D represents an initial partial response followed by disease progression. Patient E represents complete response followed by disease progression.
Limited knowledge exists on the mechanisms mediating resistance to ICIs in lung cancer. Recent research has demonstrated that some genomic alterations might confer primary resistance to ICIs, including STK11 and KEAP1 mutations [4][5]. Similarly, increased activity of enhancer of zeste homolog 2 (EZH2) has been associated with acquired resistance to ICI in NSCLC [6]. Other mechanisms, such as clonal selection of tumor cells lacking neoantigens [7], low non-synonymous tumor burden [8], and impaired HLA class I antigen function, have been described in lung cancer [9]. However, the majority of our understanding of ICI resistance comes from research done on melanoma. Although it is plausible that lung cancers share many of the pathways leading to ICI resistance with solid tumor cancers, further research is needed to clarify this. A precise understanding of the molecular underpinnings of ICI resistance pathways is needed to develop rational combination therapies that can overcome acquired ICI resistance.
2. PD-1/PD-L1 Axis in Normal Health and Tumorigenesis
The development of a naïve T cell into an effector cell involves activation of T cells in secondary lymphoid structures (such as lymph nodes) and involves three distinct signals: T cell receptor (TCR) engagement of peptide-major histocompatibility complex (MHC) (signal 1), co-stimulation between CD28 and B7-1 (CD80) or B7-2 (CD86) (signal 2), and inflammatory cytokines (such as IL-12 and type 1 interferon (IFN)) (signal 3) [10]. PD-1 is a crucial inhibitory receptor which is mainly present on activated T cells and is one of the many receptors that negatively regulate T cell activation (so-called immune checkpoints). While it is also important for fighting infections and mediating tissue repair, PD-1/PD-L1 and other checkpoint signaling is crucial for maintaining the balance between positive and negative regulation of T cells, maintaining immune tolerance, preventing autoimmunity, and avoiding excessive immune response [11].
Activated T cells produce inflammatory cytokines such as IFN-γ that lead to upregulation of PD-L1 on the antigen-presenting cells [12]. Engagement of PD-1 with PD-L1 leads to dampening of the TCR-mediated T cell activation. In addition, PD-1/D-L1 interaction leads to inhibition of the CD28-mediated co-stimulation [13]. PD-L1 can also compete with CD80 to bind to CD28, leading to further inhibition of the T cell response [14].
Despite producing numerous neoantigens (such as proteins encoded by the mutated genes [15] or aberrant transcripts produced in the dysregulated cancer cell nucleus [16]), cancer cells are often able to evade immune detection and immune-mediated killing by several mechanisms [17]. Early experiments showed that in mouse models of myeloma, tumors were found to upregulate surface PD-L1 to reduce T cell effector functions, and the growth of transplanted tumor cells could be reduced by using anti-PD-L1 antibodies [2]. Observations such as these [18][19][20], along with the findings of upregulation of PD-L1 in a variety of different solid tumors [21], led to further research and development of anti-PD-1/PD-L1 antibodies in humans. As of 2020, we now have four FDA-approved anti-PD-1 and anti-PD-L1 therapies for lung cancers alone.
3. Mechanisms of Acquired ICI Resistance
For anti-PD-1/PD-L1 therapies to be effective, several conditions need to be satisfied: (1) a sufficiently immunogenic tumor; (2) efficient activation and infiltration of T cells to the tumor microenvironment (TME); (3) tumor dependence on the PD-1/PD-L1 axis for evasion of the anti-tumor immune responses; (4) sufficient reinvigoration of exhausted tumor-specific CD8 + T cells; (5) T cell specificity for a wide variety of tumor antigens or for proteins essential for tumor growth, and display of these antigens on MHC on the surface of tumor cells; and (6) generation of long-lived effector T cells or memory T cells (durable remission). Tumors can exploit any of these steps (alone or in combinations) to acquire resistance to ICIs (Figure 2).
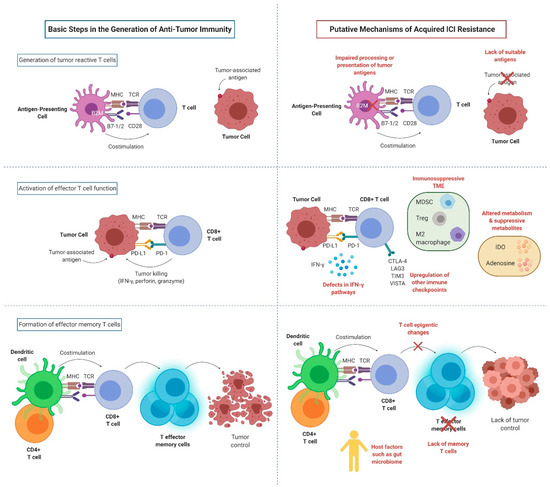
Figure 2. Key mechanisms of acquired resistance to anti-PD-1/PD-L1 inhibitors (created with BioRender.com). MHC: major histocompatibility complex; TCR: T cell receptor; CD28: cluster of differentiation 28; B7-1: cluster of differentiation 80; B7-2: cluster of differentiation 86; B2M: B2-microglobulin; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; IFN-γ: interferon gamma; CTLA-4: cytotoxic T-lymphocyte-associated protein 4; LAG3: lymphocyte-activation gene 3; TIM3: T cell immunoglobulin and mucin domain-containing molecule 3; VISTA: V-domain Ig suppressor of T cell activation; MDSC: myeloid-derived suppressor cells; Treg: regulatory T cell; IDO: indoleamine 2,3-dioxygenase.
4. Patterns of Failures After PD-1/PD-L1 Blockade
Studies examining the patterns of failures in NSCLC after PD-1/PD-L1 blockade are sparse. Difficulty in assessing progression on ICI has led to some describing acquired resistance as disease progression after partial response or complete response (CR) [22], while others have described it as initial clinical benefit followed by the development of progression [23]. Widespread use of immune-related response criteria (iRECIST) [24], which requires additional follow-up scans to confirm or refute an "unconfirmed progressive disease" designation, will hopefully harmonize the definitions of acquired resistance and lower erroneous terminations of ICI therapy. Novel methods such as circulating tumor DNA [25], TCR sequencing on peripheral T cells [26], tumor biopsies immune-profiling analyses by next-generation sequencing and high-throughput flow cytometry/mass cytometry [27], and isotope-labeled PD-1 and PD-L1 targeted imaging [28] will hopefully lead to a more accurate and reliable diagnosis of acquired ICI resistance in the future.
Patterns of failures after ICI can either be in new sites or established sites. In patients who relapse after a CR, many seem to develop new sites of metastasis, while the initial disease is controlled. In a study of ICI-treated (the majority with anti-PD-1/PD-L1 blockade) melanoma patients (N = 428), 77 patients achieved an ongoing CR, while 351 experienced progressive disease. Of the patients who survived after an incomplete response or progressive disease to ICI treatment (N = 282), 52 were treated with local therapy to control oligoprogression outside of the brain. Seven out of the eight patients (88%) who recurred after a CR recurred in a new site, which suggests that ICI can still fail to clear occult micrometastases, which can progress later as new sites of disease [29]. In contrast, progression at sites of established disease suggests that ICI was effective in clearing clinically occult micrometastases but was associated with local immune failure, likely from the development of an immune escape mechanism. Although limited by small sample size, the authors reported that the melanoma patients who achieved no evidence of disease after local therapy had significantly longer progression-free survival (PFS)when progression was limited to established tumors (N = 15) compared with patients who developed new lesions (N = 22) [29]. Similar results were seen in another study of melanoma patients treated with combined anti-PD-1 and anti-CTLA-4 therapy, with the majority of patients progressing after CR doing so in new sites versus established sites [30].
Only a handful of studies have reported the clinical outcomes of progression on PD-1 blockade therapy in NSCLC. In a study involving 34 NSCLC patients who developed acquired resistance to PD-1 axis blockade therapy, factors significantly associated with PFS were performance status and the depth of response to immunotherapy on multivariable analyses [23]. A majority of patients (61%) had progression of their existing disease rather than development of new lesions. About 2/3 of the patients progressed at a single organ, while the rest had multi-site progression. A majority of the patients (82%) had progressive disease as their best reported response to ICI [23].
In another study by Gettinger et al., 26 patients with acquired resistance to PD-1 axis inhibitor therapy were evaluated [22]. The most common site of progression was in the lymph nodes (77%). Recurrence was limited to one or two sites of disease in 88% of patients. The authors concluded that continuation of PD-1 therapy with local therapy can achieve prolonged benefit in patients with 1–2 sites of disease [22]. This observation needs to be validated in larger studies, but it is possible that prior therapy might alter the immune microenvironment or render tumors more immunogenic due to immune editing in the presence of prior chemotherapy.
5. Principles of Overcoming Acquired Resistance to PD-1/PD-L1 Blockade
Improved understanding of the underlying acquired resistance mechanisms is crucial to develop strategies to combat resistance. One can argue that ICI perhaps selectively kills only the intrinsically sensitive tumor cells, thus leaving resistant clones to persist and eventually proliferate. Mechanistic studies are thus needed to understand the highly complex and heterogeneous TME compartments that might have a differential sensitivity to T cell-mediated killing [31]. Combination therapies (such as with chemotherapy) that eliminate multiple tumor subclones from the outset can potentially prevent resistance due to the persistence of resistance clones. Another possibility for the development of acquired resistance is the immune editing and evolution of tumor and immune cells under the pressure of PD-1/PD-L1 blockade that may transform sensitive tumors into resistance tumors (as discussed in earlier sections).
Attempts to study these mechanisms will likely require the collection of sequential pre-treatment, on-treatment, and post-relapse tissue and blood samples. Pre-treatment biopsies could pave the way for biomarker-based adaptive therapy. Based on the genetic and immunological profile of the pre-treatment biopsy, either ICI alone or a "lead-in" therapy with a sensitizing agent could be instituted. Sensitizing agents could be further modified based on temporal changes on the immunological profiles of sequential biopsies and peripheral blood sampling. Evaluation of temporal changes in TME, peripheral blood chemokines, and immunological profiles has the potential to help to design adaptive therapies that can overcome acquired resistance to ICIs in an efficient way. Application of modern systems biology techniques such as computational systems biology can help to integrate the complex information gathered from tumor biopsies and peripheral blood samples into the design of effective therapeutics [32].
Various combinatorial approaches utilizing cytokines, tumor-infiltrating lymphocytes, chimeric antigen receptor T cell therapy, vaccines, chemotherapy, as well as other ICIs are currently under investigation to help to overcome resistance to PD-1 axis blockade [33]. Although an in-depth discussion of these therapeutic strategies is outside the scope of this review, we have provided a brief summary of potential therapeutic approaches to overcome ICI resistance in NSCLC (Table 1).
Table 1. Putative mechanisms and approaches to overcome acquired immune checkpoint inhibitor (ICI) resistance in non-small-cell lung cancer. Most approaches have multiple potential mechanisms of actions.
|
Mechanisms of Acquired ICI Resistance |
Potential Therapeutic Approaches |
|
Impaired processing or presentation of tumor-associated antigens |
Chemotherapy, radiotherapy, cancer vaccines, oncolytic viruses, TLR agonists |
|
Fixed epigenetic modification of T cells |
Epigenetic modulators (hypomethylating agents, histone deacetylase inhibitors) |
|
Defects in IFN-γ pathways |
STING agonists, c-di-GMP, JAK inhibitors |
|
Upregulation of other immune checkpoints |
Combination immune checkpoint inhibitors (such as CTLA-4, TIGIT, LAG-3, TIM-3, VISTA, BTLA), immune stimulatory agents (such as OX40, CD40, GITR, ICOS, 4-1BB) |
|
Immunosuppressive cells/inhibitory metabolites in the tumor microenvironment |
Macrophage inhibitors (such as CSF1R inhibitors), cytokine/chemokine inhibitors, targeted therapies targeting canonical pathways (such as against VEGF, PI3K), IDO inhibitors, TGF-β inhibitors, CXCR2 inhibitors, CXCR4 inhibitors, A2AR inhibitors/anti-CD73 |
|
Gut microbiome |
Fecal microbiota transplantation, probiotics, diet interventions |
ICI: Immune checkpoint inhibitors; TLR: Toll-like receptors; STING: Stimulator of interferon genes; c-di-GMP: Bis-(3′-5′)-cyclic dimeric guanosine monophosphate; JAK: Janus kinase; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; TIGIT: T cell immunoreceptor with immunoglobulin and ITIM domains; LAG-3: Lymphocyte-activation gene 3; TIM-3: T cell immunoglobulin and mucin-domain containing-3; VISTA: V-type immunoglobulin domain-containing suppressor of T cell activation; CD40: Cluster of differentiation 40; GITR: Glucocorticoid-induced TNFR-related; ICOS: Inducible T cell costimulatory; BTLA: B- and T-lymphocyte attenuator; CSF1R: Colony stimulating factor 1 receptor; VEGF: Vascular endothelial growth factor; PI3K: Phosphoinositide 3-kinases; IDO: indoleamine 2,3-dioxygenase; TGF-β: Transforming growth factor beta; CXCR2: CXC chemokine receptor 2; CXCR4: CXC chemokine receptor 4; A2AR: Adenosine A2A receptor; CD73: Ecto-5′-nucleotidase.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12123851
References
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800, doi:10.1038/nm73.
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297, doi:10.1073/pnas.192461099.
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer. 2018,118, 9–16, doi:10.1038/bjc.2017.434.
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835, doi:10.1158/2159-8290.cd-18-0099.
- Arbour, K.; Shen, R.; Plodkowski, A.; Rizvi, H.; Ni, A.; Long, N.; Halpenny, D.; Sanchez-Vega, F.; Rudin, C.; Riely, G.; et al. MA19.09 Concurrent Mutations in STK11 and KEAP1 is Associated with Resistance to PD-(L)1 Blockade in Patients with NSCLC Despite High TMB. J. Thorac. Oncol. 2018, 13, S424, doi:10.1016/j.jtho.2018.08.480.
- Toyokawa, G.; Takada, K.; Tagawa, T.; Hamamoto, R.; Yamada, Y.; Shimokawa, M.; Oda, Y.; Maehara, Y. A Positive Correlation Between the EZH2 and PD-L1 Expression in Resected Lung Adenocarcinomas. Ann. Thorac. Surg. 2019, 107, 393–400, doi:10.1016/j.athoracsur.2018.08.056.
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non–Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276, doi:10.1158/2159-8290.cd-16-0828.
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128, doi:10.1126/science.aaa1348.
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435, doi:10.1158/2159-8290.cd-17-0593.
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086, doi:10.1158/2159-8290.cd-18-0367.
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167, doi:10.1038/nri.2017.108.
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452, doi:10.1016/j.immuni.2018.03.014.
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1–mediated inhibition. Science 2017, 355, 1428–1433, doi:10.1126/science.aaf1292.
- Sugiura, D.; Maruhashi, T.; Okazaki, I.-M.; Shimizu, K.; Maeda, T.K.; Takemoto, T.; Okazaki, T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 2019, 364, 558–566, doi:10.1126/science.aav7062.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558, doi:10.1126/science.1235122.
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, Éric; Laverdure, J.-P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Côté, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516, doi:10.1126/scitranslmed.aau55166.
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198, doi:10.1016/j.semcancer.2015.03.004.
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096.
- Blank, C.; Brown, I.; Peterson, A.C.; Spiotto, M.; Iwai, Y.; Honjo, T.; Gajewski, T.F. PD-L1/B7H-1 Inhibits the Effector Phase of Tumor Rejection by T Cell Receptor (TCR) Transgenic CD8+T Cells. Cancer Res. 2004, 64, 1140–1145, doi:10.1158/0008-5472.can-03-3259.
- Miura, S.; Okudaira, K.; Hokari, R.; Tsuzuki, Y.; Okada, Y.; Komoto, S.; Watanabe, C.; Kurihara, C.; Kawaguchi, A.; Nagao, S.; et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int. J. Oncol. 2009, 35, 741–749, doi:10.3892/ijo_00000387.
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int Immunol 2007, 19, 813–824, doi:10.1093/intimm/dxm057.
- Gettinger, S.; Wurtz, A.; Goldberg, S.B.; Rimm, D.; Schalper, K.; Kaech, S.; Kavathas, P.; Chiang, A.; Lilenbaum, R.; Zelterman, D.; et al. Clinical Features and Management of Acquired Resistance to PD-1 Axis Inhibitors in 26 Patients With Advanced Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 831–839, doi:10.1016/j.jtho.2018.03.008.
- Shah, S.; Wood, K.; Labadie, B.; Won, B.; Brisson, R.; Karrison, T.; Hensing, T.; Kozloff, M.; Bao, R.; Patel, J.D.; et al. Clinical and molecular features of innate and acquired resistance to anti-PD-1/PD-L1 therapy in lung cancer. Oncotarget 2017, 9, 4375–4384, doi:10.18632/oncotarget.23315.
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litière, S.; Dancey, J.; Chen, A.; et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, e143–e152, doi:10.1016/s1470-2045(17)30074-8.
- Goldberg, S.B.; Narayan, A.; Kole, A.J.; Decker, R.H.; Teysir, J.; Carriero, N.J.; Lee, A.; Nemati, R.; Nath, S.K.; Mane, S.M.; et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin. Cancer Res. 2018, 24, 1872–1880, doi:10.1158/1078-0432.ccr-17-1341.
- Hogan, S.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.-F.; Nguyen-Kim, T.D.L.; et al. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2019, 7, 77–85, doi:10.1158/2326-6066.cir-18-0136..
- Auslander, N.; Zhang, G.; Lee, J.S.; Frederick, D.T.; Miao, B.; Moll, T.; Tian, T.; Wei, Z.; Madan, S.; Sullivan, R.J.; et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018, 24, 1545–1549, doi:10.1038/s41591-018-0157-9.
- Bensch, F.; Van Der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; Van Der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858, doi:10.1038/s41591-018-0255-8.
- Klemen, N.D.; Wang, M.; Feingold, P.L.; Cooper, K.; Pavri, S.N.; Han, D.; Detterbeck, F.C.; Boffa, D.J.; Khan, S.A.; Olino, K.; et al. Patterns of failure after immunotherapy with checkpoint inhibitors predict durable progression-free survival after local therapy for metastatic melanoma. J. Immunother. Cancer 2019, 7, 196, doi:10.1186/s40425-019-0672-3.
- Silva, I.C.; Long, G.V.; Quek, C.; Gonzalez, M.; Carlino, M.S.; Long, G.V.; Menzies, A.M. Site‐specific response patterns, pseudoprogression, and acquired resistance in patients with melanoma treated with ipilimumab combined with anti–PD‐1 therapy. Cancer 2020, 126, 86–97, doi:10.1002/cncr.32522.
- Williams, J.B.; Li, S.; Higgs, E.F.; Cabanov, A.; Wang, X.; Huang, H.; Gajewski, T. Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-γ-signaling mutant cancer cells. Nat. Commun. 2020, 11, 1–14, doi:10.1038/s41467-020-14290-4.
- Popel, A.S. Immunoactivating the tumor microenvironment enhances immunotherapy as predicted by integrative computational model. Proc. Natl. Acad. Sci. 2020, 117, 4447–4449, doi:10.1073/pnas.2001050117.
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164, doi:10.1200/edbk_240837.