In December 2019, the latest member of the coronavirus family, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in Wuhan, China, leading to the outbreak of an unusual viral pneumonia known as coronavirus disease 2019 (COVID-19). COVID-19 was then declared as a pandemic in March 2020 by the World Health Organization (WHO). The initial mortality rate of COVID-19 declared by WHO was 2%; however, this rate has increased to 3.4% as of 3 March 2020. People of all ages can be infected with SARS-CoV-2, but those aged 60 or above and those with underlying medical conditions are more prone to develop severe symptoms that may lead to death. Patients with severe infection usually experience a hyper pro-inflammatory immune reaction (i.e., cytokine storm) causing acute respiratory distress syndrome (ARDS), which has been shown to be the leading cause of death in COVID-19 patients. However, the factors associated with COVID-19 susceptibility, resistance and severity remain poorly understood. In this study, we thoroughly explore the correlation between various host, viral and environmental markers, and SARS-CoV-2 in terms of susceptibility and severity.
- SARS
- COVID-19
- susceptibility
- resistance
- pathogenesis
1. Introduction
In December 2019, an unusual outbreak of viral pneumonia known as coronavirus disease 2019 (COVID-19) hit Wuhan, China. COVID-19 was then declared as a pandemic in March 2020 by the World Health Organization (WHO) [1]. This novel coronavirus disease is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2); the newest addition to the coronavirus family. Coronaviruses are positive, single-stranded RNA (ssRNA) viruses that cause diseases in mammals and birds, mainly respiratory and intestinal infections [2]. There are four subgroups of coronaviruses: alpha (α), beta (β), gamma (γ) and delta (δ) [3]. In humans, they cause respiratory tract infections with severity ranging from mild to lethal. Seasonal human coronaviruses (HCoV) such as HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 contribute to approximately 15–30% of common colds [4]. In addition to the newly emerged SARS-CoV-2, two other highly pathogenic human coronaviruses have emerged in previous years; SARS-CoV (SARS) and MERS-CoV (MERS) [5]. In 2002, the outbreak of SARS started in Guangdong province, China, spreading across 26 countries in the world, affecting about 8096 individuals (9.2% fatality rate) [6]. Ten years later, the first case of MERS emerged in Saudi Arabia, leading to an ongoing endemic in the Middle Eastern region [2][3]. So far, MERS-CoV has affected 2494 individuals, with 34% fatality rate [6]. All three highly pathogenic CoV are of zoonotic origin that belong to the β subgroup [2][3].
The four main protein components of a coronavirus are spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins [7]. The name “Corona” comes from the crown-like shape of the spike protein on the outer surface of the virus [3]. This S protein is the key to the viral attachment, fusion, and entry to host cell [7]. Compared to the other human coronaviruses, SARS-CoV-2 is closely related to SARS-CoV in terms of sequence and receptor binding. The sequence of SARS-CoV-2′s S protein is approximately 76% and 80% similar to that of SARS-CoV and CoV ZXC21 (i.e., bat-like SARS-CoV), respectively [8]. Further, both SARS-CoV and SARS-CoV-2 bind to the angiotensin converting enzyme 2 (ACE2) receptors, whereas MER-CoV binds to dipeptidyl peptidase 4 (DPP4) [7][8]. ACE2 levels were found to be the highest in the small intestine, testis, kidneys, heart, thyroid, and adipose tissue, followed by medium expression in the lungs, colon, liver, bladder, and adrenal gland [9]. Additionally, enriched ACE2 expression was observed in the nose. According to a recent study, nasal epithelial cells showed the highest ACE2 expression in comparison to the other investigated respiratory cells [10]. ACE2 receptors have been also reported in oral structures, such as the tongue, the floor of the mouth and the saliva [11]. These data suggest that COVID-19 goes beyond being a respiratory disease as it may infect tissues other than the lungs. For example, several studies have reported evidence of SARS-CoV-2 in feces of COVID-19 patients [12], suggesting possible gastrointestinal infections.
SARS-CoV-2 seems to be more transmissible but less pathogenic than other zoonotic-origin CoV. The initial mortality rate of COVID-19 was declared by WHO as 2%; however, this rate has increased to 3.4% as of 3 March 2020 [1]. In fact, different fatality rates have been reported in different countries, with the lowest rate being reported in Singapore (<0.1%) and Qatar (0.2%), and highest in UK (14.8%) [13]. According to several reports, acute respiratory distress syndrome (ARDS) was shown to be the leading cause of death in severely ill COVID-19 patients [14][15][16]. Likewise, ARDS is a common immunological outcome for both SARS and MERS [17], as well as highly pathogenic influenza viruses (H5 and H7) [18]. Patients with severe COVID-19 usually experience a hyper pro-inflammatory immune reaction known as a cytokine storm, which often leads to ARDS, multiple organ failure, and eventually death [5]. Note that people of all ages can be infected with SARS-CoV-2; however, those aged 60 or above and those with underlying medical conditions are more prone to develop severe outcomes. Generally, most COVID-19 cases are actually mild or asymptomatic (80%) [1], meaning that they show mild or no symptoms at all. Based on COVID-19 figures in China, approximately four in five infected individuals are asymptomatic [19]. However, factors related to disease severity or resistance remain poorly understood. There are many extrinsic and intrinsic factors associated with COVID-19 susceptibility, resistance, and severity. These markers include viral, host, genetics, environmental, microbiome, metabolome, blood group, vitamins, and others. In this review, we thoroughly explore the correlation between such markers and SARS-CoV-2 in terms of susceptibility and severity.
2. Viral Factors
It has been well established that the human–human transmission rate of SARS-CoV-2 (R0~1.4 to 6.47) is higher than that of both MERS-CoV and SARS-CoV-1 (0.3–1.3 and 2.2–3.7, respectively) [6]. Studies have shown that the spike protein of SARS-CoV-2 harbors a furin-cleavage site (RPAR) that is absent in SARS-CoV-1 and other coronaviruses from the same clade [8][20]. Since this site is cleaved by furin contributing to S protein priming required for viral entry, this site may resemble a “gain of function” mutation, leading to a higher rate of spread in humans [8]. Further, given that furin is abundant in several tissues, it may expand the tissue tropism of SARS-CoV-2 compared to other CoV. This also applies to influenza viruses, where highly pathogenic influenza viruses contain furin-like cleavage sites leading to expansion of tissue tropism [8]. For example, H5N1 hemagglutinin A (HA) cleavage site contains a polybasic insertion (RERRRKKR↓GL), which was shown to be associated with increased virulence of the virus during the Hong Kong 1997 outbreak [21]. In addition, it is known that RNA viruses are continuously evolving, experiencing very high mutation rates that are usually associated with enhanced pathogenicity and virulence [22]. Note that antigenic drift has been observed in other coronaviruses, including SARS-CoV-1 [23]. The mutation rate of SARS-CoV-1 was estimated to be 0.80–2.38 × 10−3 substitutions per site per year [24]. On the other hand, the mutation rate of SARS-CoV-2 was estimated to be 1.05–1.26 × 10−3 substitutions per site per year, similar to that of some MERS-CoV estimates [25]. Most of the mutations occur in the surface proteins, allowing the virus to escape immune response and enhance pathogenicity [23]. So far, few mutations have been identified in circulating SARS-CoV-2 viruses, but their significance in terms of pathogenicity, transmission, and immune escape has not been identified. A mutation D614G was identified in the spike of SARS-CoV-2; this variant was first identified in Europe in February 2020 and within two months it became the most dominant variant all over the world [23]. Compared to the Wuhan reference sequence, A to G mutation is located at position 23,403 l3 leading to a change in amino acid from aspartic acid to glycine in position 614 [23]. A study has shown that this mutation is frequently found with three other mutations (i.e., transmitted as a haplotype); 241C > T in the 5′ UTR(untranslated region), a silent mutation 303C > T, and 14408C > T mutation that leads to an amino acid change in RNA-dependent RNA polymerase (P323L) [23]. In terms of structure, D614 is not located in the receptor binding domain (RBD) of S protein; rather it is located on the surface of S protein protomer forming a hydrogen bond with neighboring individual S protomers. This hydrogen bonding stabilizes the spike’s mature trimeric form on the virion surface. Thus, the change into glycine would destabilize the hydrogen bonding, possibly altering the interface protomer interactions and glycosylation patterns [23]. Through examining clinical data and SARS-CoV-2 sequences from 999 COVID-19 patients, Korber et al. (2020) identified that D614G was correlated with higher levels of viral RNA in the upper respiratory tract of patients. Additionally, their global tracking data showed that the G614 variant spreads faster than D614, suggesting a higher infectivity of G614 [23]. Based on the pseudotyping in vitro assays, G614 pseudotype virus exhibited a higher infectivity [23]. Interestingly, there was no association between D614G status and the hospitalization status (i.e., clinical severity of the disease). Together, these data suggest D614G is more infectious; however, it does not worsen the clinical outcome. On the contrary, another study has shown a strong correlation between case fatality rates and the G614 variant [26]. Based on their molecular model data, G614 stabilizes the original form of S protein (i.e., unliganded) rather than the activated form, suggesting that this form may be less infective. However, the original form of S protein plays an important role in escaping the immune response. Since this S protein is loosely bound to the receptor and the ACE2 binding site is not exposed, an immune response will not be triggered; hence, shielding the virus from anti-viral-spike antibodies [26]. Thereby, this immunological mechanism is hypothesized to be the cause for higher fatality by G614. It is worth mentioning that higher infectiousness does not always mean higher transmissibility [23], so further studies should look into the impact of G614 thoroughly in vitro and in vivo.
The SARS-CoV-2 RdRp (RNA Dependent RNA Polymerase) (also known as nsp12) has been shown to form a super complex with nsp7 and nsp8 [27]. Additionally, ExoN (Exonuclease) (nsp14) enhances the fidelity of RNA synthesis through proofreading errors made by RdRp [28]. The RdRp mutation (P323L) that is a part of the G614 haplotype has also been reported in another study, which showed that RdRp mutation at position 14480 found in Europe was associated with a higher rate of point mutations compared to that from Asia [28]. Since this mutation is located in the interface domain, possibly regulating the interaction of RdRp with other proteins, including ExoN, nsp8 and nsp7, it is hypothesized that this may contribute to an impaired proofreading ability and in turn a higher rate of mutations [28]. Nevertheless, this mutation’s impact on viral replication is yet to be studied. Another two novel mutations have been reported in nsp6 of SARS-CoV-2 at the amino acidic positions 3691 and 9659 [29]. By analyzing the structure of SARS-CoV-2 protein, including nsp6 mutation, it was shown that this mutation might favor viral infection by playing a role in viral autophagy [29]. Nonetheless, the role of autophagy in SARS-CoV-2 infection needs to be further studied in order to assess the role of the nsp6 mutation.
Collectively, all of these mutations provide potential antiviral therapeutic targets through understanding their role in viral pathogenicity and possible drug resistance. For example, the use of furin inhibitors may inhibit the process of S priming; thus, limiting the viral infection. Regarding the G614 variant, fortunately it showed sensitivity to neutralization when treated with polyclonal convalescent sera, which means antibody therapeutics are still plausible [23]. Note that the G614 status of sera used was unknown, so further experiments should be undertaken to check whether this makes a difference or not. However, higher antibody levels may be required to achieve neutralization since the preliminary results indicate that G614 is more infectious than D614 [23]. RdRp is also an important target for antiviral drugs used in COVID-19, such as remdesivir. Since the P323L mutation in RdRp is located next to a potential docking site, this raises the possibility of a potential role in drug resistance [28]. Therefore, the impact of P323L mutation on RdRp activity should be further assessed.
3. Host Factors
3.1. Statistics
Studies from Wuhan, China have found that almost 50% of the people with COVID-19 had a co-existing chronic disease (i.e., comorbidity) [30][31]. Other studies from around the globe have also reported severe symptoms of COVID-19 in individuals with underlying medical conditions. In a retrospective study of 1590 COVID-19 subjects in China, the most common comorbidity was hypertension (16.9%), followed by diabetes (8.2%) [32]. Interestingly, immunodeficiency was the lowest, accounting for only 0.2% of the subjects. In this study, it was also shown that more patients with comorbidities including hypertension, cardiovascular diseases, cerebrovascular diseases, diabetes, COPD (Chronic obstructive pulmonary disease), chronic kidney diseases and malignancy progressed to composite end-points (i.e., admission to ICU “Intensive Care Unit”, invasive ventilation or death) compared to those without. Collectively, the data showed that patients with comorbidities experienced worse clinical outcomes compared to those without. In fact, those with two or more comorbidities showed a significant increase in reaching the composite end-points compared to those with one or no comorbidity. In another study of 52 inpatients in China, death was observed in 67% of patients with comorbidities [31]. As mentioned earlier, UK has the highest mortality rate (14.8%). According to a prospective observational cohort study including 20,133 UK COVID-19 inpatients (median age ~73 years), more men were infected than women (60% vs. 40%) and overall mortality corresponded to 26% of patients [33]. The most common comorbidities reported were chronic cardiac disease (31%), uncomplicated diabetes (21%), non-asthmatic chronic pulmonary disease (18%) and chronic kidney disease (16%) [33]. Similar results were observed in another cohort study, where the main factors associated with COVID-19 death were gender (male predominance), older age and associated comorbidities including diabetes, severe asthma, cardiovascular disease and obesity [34]. Therefore, coexisting comorbidities may predispose people to adverse and poor COVID-19 clinical outcomes; this is highly dependent on the type and number of comorbidities. One possible mechanism may be immune dysregulation and inflammation induced by these diseases [35]. However, it is not yet known whether comorbidities contribute to COVID-19 susceptibility.
3.2. Diabetes
Diabetes is one of the fastest growing diseases worldwide. Even though it was established that diabetes is prevalent in COVID-19 patients and may lead to severe clinical symptoms, it is not yet verified whether it affects susceptibility to viral infection, and whether these symptoms are a direct outcome of diabetes solely or the renal and cardiovascular comorbidities usually associated with diabetes. The association between diabetes and the virus’ susceptibility/virulence is poorly understood in SARS-CoV-2, but it has been established in other coronaviruses viruses, such as MER-CoV and SARS-CoV. According to a study on MERS, more severe and prolonged lung pathology was observed in type 2 diabetic mice models [36]. This was due to the immune dysregulation including the alteration of important immune mediators such as monocytes/macrophages, CD4+ T cells, Ccl2 and Cxcl10 expression [36]. On the other hand, a study has suggested that SARS-CoV binds to ACE2 receptors in the pancreatic islets, damaging them and eventually leading to acute diabetes [37]. Here, it is the other way around where the viral infection actually causes diabetes. Similar mechanisms may also apply to SARS-CoV-2 infection, considering that both viruses use the same receptor. This phenomenon was also observed with influenza A viruses, where they were shown to be able to infect human pancreatic cells as well as induce pancreatic damage in animal models (in vivo) leading to diabetes [38]. As a result, hyperglycemia may lead to immune imbalance, including impaired monocyte/macrophage functions and pro-inflammatory cytokine productions [39], which may contribute to COVID-19′s severity. In contrast, hypoglycemia was reported at least once in approximately 10% of COVID-19 patients with type 2 diabetes [40]. Hypoglycemia has been associated with pro-inflammatory monocytes’ mobilization and enhanced platelet reactivity [39]. Thus, it is not yet clear whether hyperglycemia or hypoglycemia leads to poor clinical outcomes in COVID-19 patients.
3.3. Obesity and Obesogenic Comorbidities
Obesity is a global epidemic that causes a low-grade chronic inflammation, affecting the immune system. The effects include immune response dysfunction, gut microbiome/virome imbalance, pro-inflammatory responses and antiviral immunity reduction [41]. A case control study in Mexico has found that obesity predisposes COVID-19 with the strongest association, followed by diabetes and hypertension [42]. In terms of severity, a study of 30 COVID-19 subjects has reported that patients with a higher BMI (Body Mass Index) experienced more severe symptoms in comparison to those with lower BMI (27.0 ± 2.5 vs. 22.0 ± 21.3) [43]. Excess adiposity caused by obesity may lead to various chronic diseases such as diabetes and hypertension [44]. These obesogenic comorbidities affect the renin–angiotensin system resulting in metabolic imbalance and excess pro-inflammatory response [41]; as a consequence, obese patients with COVID-19 may experience severe symptoms. Obesity is also associated with dysregulation in the production of adipokines (i.e., cytokines secreted by adipose tissue) [44]. For instance, serum amyloid-A acts directly on macrophages, facilitating the increase in inflammatory cytokines secretion, including IL-6 [45], which is an important component of the cytokine storm; commonly observed in severely ill COVID-19 patients. Collectively, these data suggest a possible mechanism in which obesity may influence COVID-19′s clinical severity.
3.4. Roles of Host Enzymes
The S glycoprotein is a class I fusion protein that mediates a dual role in the infection process: binding to receptor and fusion with the host membrane. This process is mediated by three main enzymes on the host cells: ACE2, TMPRSS2, and furin. Hence, variants of these enzymes and their expression profiles might play a crucial role in the prognosis of COVID-19 patients.
The proteolytic cleavage of the spike protein at the cleavage site enables its conformational change for virus internalization to host cell [5]. This cleavage is mediated by furin, a protease readily expressed in lung cells. Another readily expressed protease, TMPRSS2, accounts for the spike protein priming as well. On the other hand, studies have shown that ACE2 expression is significantly upregulated in lung tissues of severe COVID-19 patients with comorbidities compared to the control group [46]. ACE2 upregulation is positively correlated with genes involved in histone modifications, such as HAT1, HDAC2 and KDM5B [46]. Hence, it is hypothesized that histone modification (i.e., epigenetic regulation) may contribute to ACE2 upregulation and hence SARS-CoV-2 infection. It is worth mentioning that TMPRSS2 and furin were highly expressed in lungs, but not differentially expressed across lung transcriptomes from COVID-19 patients with comorbidities and the control group. These data suggest that ACE2 may act as a limiting factor for SARS-CoV-2 infection. Therefore, ACE2 upregulation correlates with a higher possibility of severe COVID-19 through mediating SARS-CoV-2 entry into the lung cells.
4. Genetics
4.1. Overview
Population genetics have been widely associated with susceptibility and resistance to infectious diseases, including viral infections. Geographical variations of COVID-19 have been reported, where the highest rate of infections was observed in Europe (1,544,145) and the lowest in Africa (30,536) [47]. Nevertheless, African Americans correspond to 43% of COVID-19 deaths in the US [48]. A recently published review proposed that the high frequency of p.Ser1103Tyr-SCN5A variant in African Americans makes them susceptible to ventricular arrhythmia (VA) and sudden cardiac death (SCD) induced by COVID-19 (i.e., an intrinsic genetic susceptibility influenced by COVID-19 risk factors including hypoxemia and cytokine storm) [48]. In addition, the Italian-Spanish genome wide association studies (GWAS) identified susceptibility loci at chromosome 3p21.31 with a cluster of several genes associated with respiratory failure in COVID-19 [49]. This risk allele was found at a higher frequency in severe patients requiring oxygen ventilators, suggesting a possible contribution to COVID-19 severity. So, how do the host genetics come into play? It is not yet clear whether it is genetics, social factors and/or a combination of both that contribute to the current geographical variations of COVID-19. Currently, a global database (The COVID-19 Host Genetics Initiative) has been developed to identify genetic factors of COVID-19 in terms of susceptibility and severity (https://www.covid19hg.org/).
4.2. ACE2 Gene
The ACE2 gene, located in the X chromosome, is characterized by single nucleotide polymorphisms (SNPs) in the coding region, leading to different allele variants with varying frequencies among different populations [47]. For example, K31R and Y83H protective variants of ACE2 are observed with higher frequencies in Asian populations, whereas those of European descent show a higher frequency of T921 risk variant. In fact, using the S-protein-interacting synthetic mutant map of ACE2, a study has identified natural ACE2 variants that may possibly provide resistance against SARS-CoV-2 infection [50]. These variants include K31R, N33I, H34R, E35K, E37K, D38V, Y50F, N51S, M62V, K68E, F72V, Y83H, G326E, G352V, D355N, Q388L and D509Y. Therefore, the ACE2 polymorphism can affect SARS-CoV-2 susceptibility since the protective ACE2 variants showed diminished binding to the spike protein compared to risk variants. Comorbid conditions associated with COVID-19, such as diabetes and hypertension, are also modulated by ACE2 and the renin–angiotensin system as discussed previously (comorbidity section). Rather than solely being a receptor, ACE2 modulates the downstream inflammatory pathways post-infection [47]. Taken together, ACE2 may play a role in the severity of clinical outcomes in addition to its role in susceptibility to SARS-CoV-2 infection.
In terms of epigenetic regulation, a study has shown that ACE2 expression can be regulated by DNA methylation in lupus patients [51]. Lupus is an autoimmune disease where the body attacks itself through its hyper-immune system. Hypomethylation of ACE2 was observed in CD4+ T cells, leading to an overexpression of ACE2 in lupus patients compared to the healthy controls. Therefore, oxidative stress induced by COVID-19 in combination with DNA methylation deficiency in lupus patients leads to ACE2 overexpression by inducing hypomethylation at the epigenetic level. In addition, hypomethylation of interferon genes, including NFkB, has been observed. This may correlate with an increase in the cytokine storm. Collectively, these modifications will enhance SARS-CoV-2 entry into lupus patients, increasing their susceptibility to COVID-19. Note that the study did not include data from alveolar epithelial cells in lupus patients. Nevertheless, ACE2 overexpression in immune cells may contribute to SARS-CoV-2 susceptibility and cytokine storm induced organ damage in COVID-19 patients. This phenomenon may also apply to individuals with other comorbidities, explaining why ACE2 is overexpressed in such individuals. As mentioned previously, ACE2 overexpression was correlated with the upregulation of genes involved in histone modifications, such as HAT1, HDAC2 and KDM5B, in individuals with comorbidities [46]. Therefore, a combinational effect of hypomethylation and histone modifications may upregulate ACE2 expression; thus, increasing susceptibility to SARS-CoV-2 infection (Figure 1). Note that there are two isoforms of human ACE2, a full-length transmembrane protein (UniProt ID: Q9BYF1-1, 805 amino acids) and a smaller soluble isoform (UniProt ID: Q9BYF1-2, 555 amino acids) [52]. Since SARS-CoV-2 favors and binds to the membrane-bound ACE2, further studies should look into how epigenetics and the ACE2 polymorphism may account for different isoforms of ACE2, possibly leading to an increased susceptibility or resistance to SARS-CoV-2 infection.
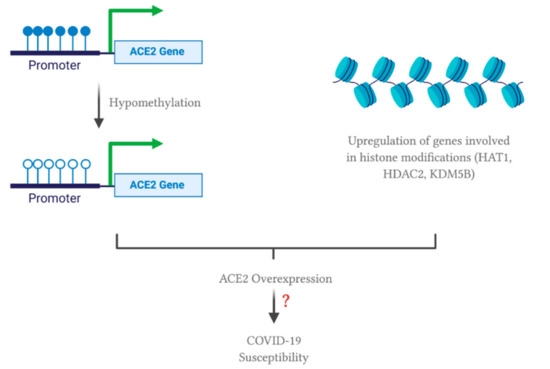
Figure 1. Possible effect of comorbidities on epigenetic regulation of angiotensin converting enzyme 2 (ACE2). All of the figures were created with BioRender.com.
4.3. Immune System Related Genes
Regulation of immune related genes may contribute to COVID-19 susceptibility. Human leukocyte antigens (HLA) are proteins encoded by the major histocompatibility complex (MHC) that allow the immune system to differentiate between self and non-self cells. They are characterized by extreme diversity and polymorphisms, accounting for susceptibility against several infectious diseases. In terms of SARS-CoV-2, an in silico study has found that individuals with HLA-B*46:01 variants may be susceptible to COVID-19, whereas HLA-B*15:03 represents a protective variant since it could provide T-cell based immunity [53]. Interestingly, the susceptible allele, HLA-B*46:01, originated in South East Asia; on the other hand, the East Asian gene pool completely lacks the protective allele, HLA-B*15:03 [47]. Therefore, the correlation between HLA variants and COVID-19 needs to be further studied to pinpoint the effect of genetic factors. In relation, a study of sequence analysis has identified 22 variants in the coding regions of some proteases (FURIN, PLG, PRSS1, TMPRSS11a) and innate immune-related genes (MBL2 and OAS1) in a Serbian population [54]. Using in silico analyses, 10 of these variants were predicted to be protein-altering variants, possibly affecting the protein’s function. For example, proteases are involved in proteolytic cleavage of the spike protein, so these variants may provide a “gain of function” mutation enhancing the proteases’ activity. On the other hand, the mutations in innate immune-related genes are hypothesized to be disadvantageous to the host, allowing the virus to escape the immune response. These variants include p.Gly146Ser in FURIN; p.Arg261His and p.Ala494Val in PLG; p.Asn54Lys in PRSS1; p.Arg52Cys, p.Gly54Asp and p.Gly57Glu in MBL2; p.Arg47Gln, p.Ile99Val and p.Arg130His in OAS1 [54]. Additional population genetics studies have shown that seven variants in PLG, TMPRSS11a, MBL2 and OAS1 genes experienced genetic divergence (i.e., different allelic frequencies) among different populations worldwide [54]. It is also interesting to note that cytokine secretion is modulated through genetics and epigenetics regulations. Even though ethnicity has been found to influence the distribution and polymorphisms of cytokines related genes [47], the effect of cytokine gene polymorphisms on SARS-CoV-2 infection has not been studied yet.
This entry is adapted from the peer-reviewed paper 10.3390/v13010045
References
- Coronavirus Disease (COVID-19) Situation Reports. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 25 June 2020).
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192.
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020.
- Ahn, D.G.; Shin, H.J.; Kim, M.H.; Lee, S.; Kim, H.S.; Myoung, J.; Kim, B.T.; Kim, S.J. Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). J. Microbiol. Biotechnol. 2020, 30, 313–324.
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108.
- Tang, D.; Comish, P.; Kang, R. The hallmarks of COVID-19 disease. PLoS Pathog. 2020, 16, e1008536.
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620.
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742.
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 1–7.
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687.
- Azim, A.A.; Shabbir, J.; Khurshid, Z.; Zafar, M.S.; Ghabbani, H.M.; Dummer, P.M.H. Clinical endodontic management during the COVID-19 pandemic: A literature review and clinical recommendations. Int. Endod. J. 2020, 53, 1461–1471.
- Xiao, F.; Sun, J.; Xu, Y.; Li, F.; Huang, X.; Li, H.; Zhao, J.; Huang, J.; Zhao, J. Infectious SARS-CoV-2 in feces of patient with severe COVID-19. Emerg. Infect. Dis. 2020, 26, 1920.
- COVID-19 Mortality Analyses, John Hopkins. Available online: https://coronavirus.jhu.edu/data/mortality (accessed on 25 June 2020).
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422.
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; Yu, T.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481.
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the alert for cytokine storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063.
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613.
- Sutton, T.C. The pandemic threat of emerging H5 and H7 avian influenza viruses. Viruses 2018, 10, 461.
- Day, M. Covid-19: Four fifths of cases are asymptomatic, China figures indicate. BMJ 2020, 369, m1375.
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6.
- Claas, E.C.; Osterhaus, A.D.; Van Beek, R.; De Jong, J.C.; Rimmelzwaan, G.F.; Senne, D.A.; Krauss, S.; Shortridge, K.F.; Webster, R.G. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 1998, 351, 472–477.
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003.
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827.e19.
- Zhao, Z.; Li, H.; Wu, X.; Zhong, Y.; Zhang, K.; Zhang, Y.P.; Boerwinkle, E.; Fu, Y.X. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol. Biol. 2004, 4, 21.
- SARS-CoV-2 Genetics, John Hopkins Center for Health Security. Available online: https://www.centerforhealthsecurity.org/resources/COVID-19/COVID-19-fact-sheets/200128-nCoV-whitepaper.pdf (accessed on 10 July 2020).
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pract. 2020, 74, e13525.
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of RNA-dependent RNA polymerase from 2019-nCoV, a major antiviral drug target. BioRxiv 2020.
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 1–9.
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27.
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513.
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062.
- Guan, W.J.; Liang, W.H.; Zhao, Y.; Liang, H.R.; Chen, Z.S.; Li, Y.M.; Liu, X.Q.; Chen, R.C.; Tang, C.L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with Covid-19 in China: A Nationwide Analysis. Eur. Respir. J. 2020, 55, 2000547.
- Docherty, A.B.; Harrison, E.M.; Green, C.A.; Hardwick, H.E.; Pius, R.; Norman, L.; Holden, K.A.; Read, J.M.; Dondelinger, F.; Carson, G.; et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: Prospective observational cohort study. BMJ 2020, 369, m1985.
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436.
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 1–12.
- Kulcsar, K.A.; Coleman, C.M.; Beck, S.E.; Frieman, M.B. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS-CoV infection. JCI Insight 2019, 4, e131774.
- Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Binding of SARS coronavirus to its receptor damages islets and causes acute Diabetes. Acta Diabetol. 2010, 47, 193–199.
- Capua, I.; Mercalli, A.; Pizzuto, M.S.; Romero-Tejeda, A.; Kasloff, S.; De Battisti, C.; Bonfante, F.; Patrono, L.V.; Vicenzi, E.; Zappulli, V.; et al. Influenza A viruses grow in human pancreatic cells and cause pancreatitis and diabetes in an animal model. J. Virol. 2013, 87, 597–610.
- Hussain, A.; Bhowmik, B.; do Vale Moreira, N.C. COVID-19 and diabetes: Knowledge in progress. Diabetes Res. Clin. Pract. 2020, 162, 108142.
- Zhou, J.; Tan, J. Diabetes patients with COVID-19 need better blood glucose management in Wuhan, China. Metabolism 2020, 107, 154216.
- Petrakis, D.; Margină, D.; Tsarouhas, K.; Tekos, F.; Stan, M.; Nikitovic, D.; Kouretas, D.; Spandidos, D.A.; Tsatsakis, A. Obesity‑a risk factor for increased COVID‑19 prevalence, severity and lethality. Mol. Med. Rep. 2020, 22, 9–19.
- Hernández-Garduño, E. Obesity is the comorbidity more strongly associated for Covid-19 in Mexico. A case-control study. Obes. Res. Clin. Pract. 2020, 14, 375–379.
- Liu, M.; He, P.; Liu, H.G.; Wang, X.J.; Li, F.J.; Chen, S.; Lin, J.; Chen, P.; Liu, J.H.; Li, C.H. Clinical characteristics of 30 medical workers infected with new coronavirus pneumonia. Zhonghua Jie He He Hu Xi Za Zhi= Zhonghua Jiehe He Huxi Zazhi= Chin. J. Tuberc. Respir. Dis. 2020, 43, E016.
- Ignacio, R.M.; Kim, C.S.; Kim, S.K. Immunological Profiling of Obesity. J. Lifestyle Med. 2014, 4, 1–7.
- Evans, A.C.; Papachristou, G.I.; Whitcomb, D.C. Obesity and the risk of severe acute pancreatitis. Minerva Gastroenterol. Dietol. 2010, 56, 169–179.
- Pinto, B.G.; Oliveira, A.E.; Singh, Y.; Jimenez, L.; Gonçalves, A.N.A.; Ogava, R.L.; Creighton, R.; Peron, J.P.S.; Nakaya, H.I. ACE2 expression is increased in the lungs of patients with comorbidities associated with severe COVID-19. MedRxiv 2020.
- Debnath, M.; Banerjee, M.; Berk, M. Genetic gateways to COVID-19 infection: Implications for risk, severity, and outcomes. FASEB J. 2020, 34, 8787–8795.
- Giudicessi, J.R.; Roden, D.M.; Wilde, A.A.; Ackerman, M.J. Genetic susceptibility for COVID-19–associated sudden cardiac death in African Americans. Heart Rhythm 2020, 17, 1487–1492.
- Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; Asselta, R.; et al. The ABO blood group locus and a chromosome 3 gene cluster associate with SARS-CoV-2 respiratory failure in an Italian-Spanish genome-wide association analysis. MedRxiv 2020.
- Stawiski, E.W.; Diwanji, D.; Suryamohan, K.; Gupta, R.; Fellouse, F.A.; Sathirapongsasuti, F.; Liu, J.; Jiang, Y.P.; Ratan, A.; Mis, M.; et al. Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. BioRxiv 2020.
- Sawalha, A.H.; Zhao, M.; Coit, P.; Lu, Q. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin. Immunol. 2020, 215, 108410.
- Tabish, M. Alternative splicing of ACE2 possibly generates variants that may limit the entry of SARS-CoV-2: A potential therapeutic approach using SSOs. Clin. Sci. 2020, 134, 1143–1150.
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human leukocyte antigen susceptibility map for SARS-CoV-2. J. Virol. 2020.
- Klaassen, K.; Stankovic, B.; Zukic, B.; Kotur, N.; Gasic, V.; Pavlovic, S.; Stojiljkovic, M. Functional prediction and comparative population analysis of variants in genes for proteases and innate immunity related to SARS-CoV-2 infection. BioRxiv 2020.