Viruses are opportunistic intracellular pathogens that are dependent on the host for their replication. They hijack host cellular machinery for their replication and survival by targeting crucial cellular physiological pathways, including transcription, translation, immune pathways, and apoptosis. Immediately after translation, the host and viral proteins undergo a process called post-translational modification (PTM). PTMs of proteins involves the attachment of small proteins, carbohydrates/lipids, or chemical groups to the proteins and are crucial for protein's functioning. During viral infection, host proteins utilize PTMs to control the virus replication, using strategies like activating immune response pathways, inhibiting viral protein synthesis, and ultimately eliminating the virus from the host. PTM of viral proteins increases solubility, enhances antigenicity and virulence properties. However, RNA viruses are devoid of enzymes capable of introducing PTMs to their proteins. Hence, they utilize the host PTM machinery to promote their survival. Proteins from viruses belonging to the family: Togaviridae, Flaviviridae, Retroviridae, and Coronaviridae such as chikungunya, dengue, zika, HIV, and coronavirus are a few well-known to be modified. This review discusses various host and virus-mediated PTMs that play a role in the outcome during the infection.
- RNA viruses
- post-translation modification
- pathogenesis
- ubiquitination
- acetylation
- glycosylation
- phosphorylation
- ISGylation
- ADP ribosylation
- SUMOylation
- PTMs
1. Introduction
Viruses are intracellular pathogens that infect a wide range of organisms, from single-cell bacteria to multicellular organisms like plants and animals. Most of the RNA viruses have a small genome (typically <14kb) [1], while coronavirus, toroviruses, and roniviruses have larger genome sizes (24-30kb) [1][2]. Regardless of the genome sizes, viruses depend on the host cell machinery for various biological and physiological functions associated with their genome replication, transcription, translation, packaging, and release from infected cells.
Post-translational modifications (PTMs) of proteins are essential for cellular housekeeping functions of the cells. PTMs include adding small proteins or functional groups such as ubiquitination, lipidation, glycosylation, methylation, phosphorylation, and acetylation to specific amino acids within the protein [3][4][5]. PTMs are carried out by specialized enzymes, such as ubiquitin E3 ligase, glycosyltransferase, poly (Adenosine diphosphate) ribose polymerase (PARP), acetyltransferase, and kinases [3][6]. PTMs enhance protein solubilization, conformation (by altering charge or hydrophobicity), interactions, signaling, degradation, and thus play a crucial role in cell growth [7]. release, and interferon response inhibition. In contrast, the host overcomes viral infection by inactivating viral proteins by either removing PTMs, which are crucial to the enzymatic activity of viral proteins or attaching small molecules such as ubiquitin or ubiquitin-like proteins leading to their inactivation and/or proteasomal mediated degradation. This review discusses some essential PTMs associated with host-pathogen interplay related to RNA viruses. Based on the size/chemical nature, PTMs can be broadly categorized into four categories: (1) Protein-based modification involving ubiquitin (known as ubiquitination), SUMO (Small ubiquitin-like modifier, known as SUMOylation), ISG (Interferon-stimulated gene, known as ISGylation), NEDD8 (Neural precursor cell expressed, developmentally down-regulated 8, known as NEDDylation), (2) carbohydrate molecule-based modification such as glycosylation, ADP ribosylation, (3) lipid molecule-based modifications such as palmitoylation, myristoylation, prenylation, and (4) chemical/ionic group such acetyl, phosphate, and methyl to the nascent proteins (Figure 1).
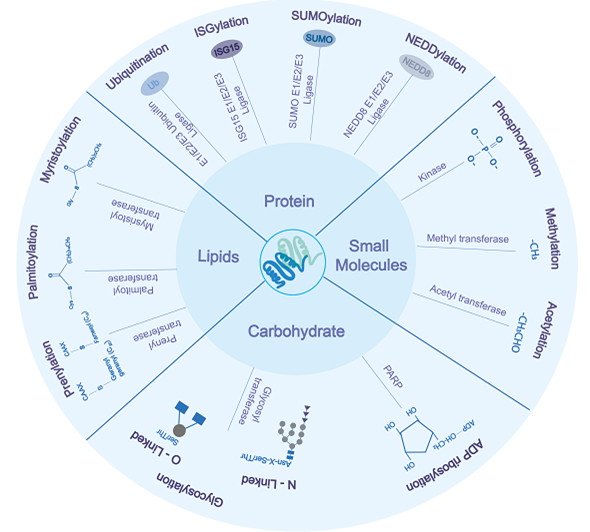
Figure 1. Overview of various post-translational modifications of virus and host proteins having an important role during viral infections. Based on the biochemical nature of attached moieties, post-translational modification (PTMs) can be categorized into modification by small protein groups (ubiquitination, ISGylation, SUMOylation, NEDDylation), carbohydrates moieties (glycosylation, ribosylation), lipids (palmitoylation, myristoylation, and prenylation), and small chemical groups (phosphorylation, methylation, and acetylation). The proteins upon modification result in loss/gain in function, which modulates the virus’s life cycle and host response to virus infection.
2. Protein-Based PTMs
Proteins such as ubiquitin and ubiquitin-like proteins (Ubls) including SUMO (Small ubiquitin-like modifier), NEDD8 (Neuronal precursor cell-expressed developmentally down-regulated protein 8), ISG15 (Interferon-stimulated gene 15), FAT10 (Human leukocyte antigen F locus adjacent transcript 10), URM1(Ubiquitin-related modifier-1), UFM1 (Ubiquitin-fold modifier 1) and ATG8 (Autophagy-related protein 8) are attached to the host and viral proteins involving activating enzyme E1, ubiquitin-conjugating enzyme E2 and ubiquitin ligase E3[8]. E1 activates the ubiquitin or ubiquitin-like proteins and then transfers them to E2, which then transfers the molecule to the protein substrate via the activity of E3 ligase. These small proteins have a similar 3D structure like that of ubiquitin (β1 β2 α1 β3 β4 α2 β5), but lack sequence similarity and are called ubiquitin-like proteins[9]. The size of these PTMs varies from 8 kDa for ubiquitin and 17 kDa for ISG15, as they have other peptide chains in addition to the ubiquitin-like domain. Among the ubiquitin-like protein modification, the role is well-known in ubiquitin, SUMO, NEDD8, and ISG15 during the viral infection, while for others such as FAT10, URM1, UFM1, and ATG, very few reports are available. These modifications act as anti-viral by either activating immune response or targeting the viral proteins to degradation or act as pro-viral by downregulating immune response or activating enzymatic/functional activities of viral proteins (Figure 1). We describe below the various ubiquitin-like modifications and their role during virus infection.
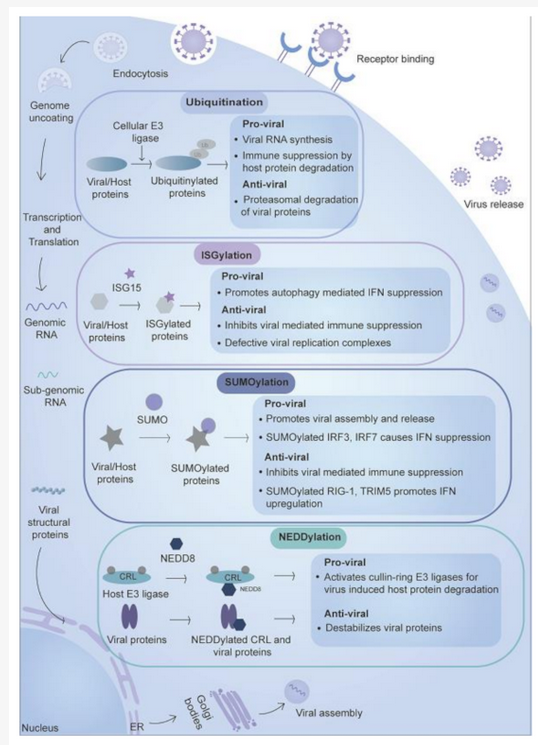
Figure 2. Small protein-based post-translational modifications during viral infection. Ubiquitin and ubiquitin-like molecules such as ISG15, NEDD8, and SUMO play an essential role during viral infection. Specific enzymes such as E1, E2, and E3 are involved in modification. The anti-viral effect is exerted by inducing IFN response, destabilizing viral proteins, or targeting viral proteins to their degradation. These modifications also exert a pro-viral impact by improving the enzymatic activity or interactions of the viral proteins, which promotes viral replication and degradation of host proteins involved in immune response proteins.
2.1. Ubiquitination
Ubiquitination is a vital modification that plays a role in various processes and maintains homeostasis under normal physiological conditions and disease/infections [10]. It involves covalent attachment of ubiquitin-protein (of around 76 amino acids long) to C-terminal glycine residue of target protein via lysine amino acid of ubiquitin (Ub) known as the canonical pathway. The other pathway, called a non-canonical pathway, involves cysteine, serine, and threonine to a target protein [11][12]. These modifications play both pro-viral and anti-viral roles during viral infections and are described below.
2.1.1. Role of Ubiquitination in the Inhibition of Viral Replication
As an innate mechanism to counter viral infection, the host performs ubiquitination of viral proteins and targets them for degradation, limiting the viral spread (Figure 2) [13][14]. Various ubiquitin ligases such as TRIM5a (Tripartite motif-containing 5a) [15], TRIM24 [16], TRIM33 [17], TRIM41 [18], and E6AP, also identified as Ubiquitin-protein ligase (E3A) [19], are known to ubiquitinate viral proteins. Ubiquitination during viral infection negatively affects the viral replication by:
(i) Tagging the proteins with Ub and targeting them for proteasomal degradation, e.g., ubiquitination of Dengue (DENV) NS3 results in its proteolytic degradation [13]. DENV NS3 forms a protease complex (NS2B3) with NS2B and cleaves cGAS (cyclic GMP-AMP synthase) and STING (Stimulator of Interferon Genes) [20][21]. Thus, degradation of NS3 negatively affects viral replication by inhibiting the protease complex formation (which is needed for viral protein processing) and increasing the interferon response due to less degradation of STING and cGAS. Nucleoprotein (NP) of Influenza, a virus (IAV), and vesicular stomatitis virus (VSV) are targeted for ubiquitin-mediated proteasomal degradation. As NP is a structural component of the negative-sense RNA virus genome, a low level of nucleoprotein allows host machinery to degrade viral RNAs, thereby limiting viral infection[18][22]. Other examples include HIV (Human immunodeficiency virus) integrase (IN) protein, which is polyubiquitinated and targeted to proteasomal degradation and affects viral replication as well as pro-viral DNA formation [17][19]. HCV (Hepatitis C virus) core protein, which is also ubiquitinated and targeted to degradation [19] thus negatively affecting the crucial viral processes such as replication, assembly as well as a counter-response to the interferon-mediated attack on the virus (Figure 2).
(ii) Inhibition of complex viral formation leading to a reduction in viral replication. DENV NS1 forms a replication complex with NS4B (non-structural 4B). Its ubiquitination prevents its association with NS4B (non-structural 4B) and promotes its oligomerization, thus preventing replication complex formation [23], leading to restricted viral replication. Besides ubiquitinating viral proteins, E3 ligases auto ubiquitinate various TRIM (Tripartite motif) proteins. A few examples are TRIM5a [14] and TRIM26 [24], which lead to enhanced expression of interferons or degradation of viral proteins, thus protecting the host.
The ubiquitination of viral proteins in some cases promotes viral replication through various mechanisms such as (i) ubiquitin-dependent enzymatic activity of viral proteins, e.g., Ebola virus protein VP35, which is a cofactor of the viral polymerase as well as an inhibitor of the host anti-viral type I interferon (IFN-I) system [6], PB2 (Polymerase basic protein 2) (which enhances polymerase action and thus viral replication), nucleoprotein of influenza virus (promote replication and assembly) [25][26][27], and HCV NS2 (which promotes viral assembly) [28]. Ubiquitination of the polymerase subunit of these viruses positively affects enzymatic activity, thus promoting viral replication (Figure 2).
(ii) Ubiquitin-mediated negative effect on the host immune response leading to a positive effect on viral replication, e.g., Zika virus (ZIKV) NS1 interacts with USP8 (Ubiquitin Specific Peptidase 8) and causes the deubiquitination of Caspase 1 and prevents its proteasomal degradation. Enhanced stabilization of Caspase 1 promotes the cleavage of cGAS, which is involved in the initiation of IFN-I signaling [29]. NS5 of ZIKV and yellow fever viruses bind to and prevent the ubiquitination of RIG-1 (Retinoic acid-inducible gene 1), followed by inhibition of both phosphorylation and nuclear translocation of IRF3 (IFN regulatory factor 3) [30], leading to the suppression of IFN response. ZIKV NS3 and NS2B3 bind to host MAVS (Mitochondrial anti-viral signaling protein) and MITA (Mediator of IFN regulatory factor 3 activation) and target them for degradation by catalyzing their K48-linked ubiquitination [31].
(iii) Host and viral encoded deubiquitinating enzymes remove ubiquitin and promote viral growth. Viruses negatively affect innate immune response by deubiquitinating host immune pathways proteins [32] e.g., SARS-CoV (Severe acute respiratory syndrome coronavirus) and MERS-CoV (Middle east respiratory syndrome coronavirus) encoded protein such as PLpro (papain-like protease) exhibit deubiquitinating enzymatic activity and deISGylating activity and affect cytokine and interferon response [33][34] by reducing the expression of CCL5 (Chemokine ligand 5), CXCL10, (C-X-C motif chemokine ligand 10) and IFN-β [35]. MERS-PLpro displays broad cleavage specificity towards poly-Ub chains and suppresses the IFNβ signaling pathway [36]. SARS-PLpro cleaves K48 (preferentially) and K63 -linked poly-Ub chains of Tumor necrosis factor receptor (TNFR)-associated factor 3 (TRAF3) and TRAF6, thereby inhibiting NF-kB (Nuclear factor kappa B) activation [37], whereas SARS-CoV-2-PLpro have a preference for ISG15 as compared to ubiquitin [34][38]. Additionally, host deubiquitinating enzymes USP7 promotes HIV replication by removing ubiquitin from Tat (Trans-Activator of Transcription) protein, which enhances the viral transcription efficiency, rescuing it from degradation [39]. These examples show that both ubi and deubiquitination play an important role in regulating immune response and viral growth.
2.2. ISGylation
Viral infections induce an immune response, an anti-viral state in cells by inducing expression of IFN-I production downstream of the JAK-STAT (Janus kinase-signal transducer and activator of transcription) pathway leading to increased transcription of ISGs (Interferon-stimulated genes) [40] Microarray studies suggest around 300 ISGs are involved in various anti-viral defense functions. These include but are not limited to cellular signaling, inflammation, transcription activation, antigen processing, and immune modulation [41]. Among the ISGs, ISG15 plays a role in PTM. In contrast, GBP1 (Guanylate-binding protein 1), IFIT1 (Interferon Induced Protein With Tetratricopeptide Repeats 1), IFIT2, ISG20, ZAP (zinc finger anti-viral protein) bind to viral RNA or inhibit apoptosis [42]. Here we will discuss the anti-viral and pro-viral actions of ISG15 as PTM, as shown in Figure 2.
2.2.1. Role of ISGylation in the Inhibition of Viral Replication
ISG15 conjugation plays an anti-viral role during viral infection by conjugation to viral proteins or hosts cellular proteins, leading to inhibition of enzymatic activity, viral replication inhibition, translational shutoff, and metabolic reprogramming [43][44]. ISGylation inhibits the viral cycle by (i) targeting viral replication and release. ISG15 interferes with Ebola VP40 (matrix protein) mediated viral egress. It targets and negatively affects Nedd4 ligase, which ubiquitinates VP40 and is helpful for the VP40 activity. ISGylation leads to a reduction in the viral release [43]. Influenza B virus (IBV) viral genome occurs in the form of ribonucleoprotein complex (RNP). The RNP assemblies facilitate IBV RNA synthesis and comprise a viral polymerase, viral RNA, and NP oligomers. During viral infection, ISGylation of viral NP blocks viral RNA synthesis by inhibiting oligomerization leading to reduction of viral RNA synthesis. NS1B reduces modified NP by sequestering to minimize the detrimental effect ISGylation; however, the dominant-negative effect (a minute fraction of modified NP left from NS1B sequestration) negatively affects viral RNA synthesis [45] (Figure 2).
(ii) Enhancing the interferon response. ISGylation induced anti-viral in non-hematopoietic cells during Coxsackievirus B3 (CVB3) infection. The authors suggest that ISG conjugation to anti-viral effectors IFIT1 and IFIT3 blocks ubiquitination-dependent degradation and stabilizes these proteins leading to an enhanced anti-viral response [46] (Figure 2). CVB3 encodes a protease (2APro) that mediates host cell translational shut off by cleavage of eukaryotic translation factor 4γ1 (eIF4G1), which binds to RNA cap involved in cap-dependent translation. The eIF4G1 degradation favors the viral RNA translation as the CVB3 genome lacks cap and is translated in a cap-independent manner. However, ISGylation of 2APro impairs the eIF4G1 cleavage during CVB3 infection, causing suppression of viral replication [4].
2.2.2. Role of ISGylation in Promotion of Viral Replication
ISGylation is a part of the anti-viral defense system, which helps in controlling viral replication. In some cases, the modification serves the pro-viral function for the virus via inhibition of the immune pathways. During RNA virus infection, LRRC25 (Leucine-rich repeat-containing protein 25) interacts with ISG15 modified RIG-1 and promotes degradation via autophagy in a p62-dependent manner, thereby downregulating IFN response [47] (Figure 2). Studies with SARS-CoV2-PLpro found that it blocks the IRF3 pathway (by inhibiting the phosphorylation of IRF3) and NF-kB pathway (by an unknown mechanism) [38][48].
2.3. SUMOylation
SUMOylation requires conjugation of a small ubiquitin-like modifier (SUMO) to a lysine residue in the consensus sequence ΨKxD/E within the target protein (Ψ represents hydrophobic amino acid residue, x is any amino acid, D/E is an acidic amino acid). Proteins can be mono- or poly-sumoylated [49]. SUMOylation regulates critical cellular processes such as transcriptional regulation, DNA repair, and innate immunity [9]. Covalently attached SUMO moieties regulate protein stability, localization, and interactions by bringing about conformational changes in target proteins [50]. In addition to covalently linked SUMO chains, proteins with a SUMO-interaction motif (SIM) can interact with SUMO moiety non-covalently. Such non-covalent interactions assist in ubiquitination due to the SIM motif in SUMO-targeted ubiquitin ligases (STUbLs) [51]. SUMOylation can inhibit viral replication by promoting immune response and act as a pro-viral by tagging host proteins, which hampers IFN response (Figure 2).
2.3.1. Role of SUMOylation in the Inhibition of Viral Replication
Similar to ubiquitination, SUMOylation can inhibit viral replication. SUMO conjugation to virally encoded proteins is implicated in stabilization and pertains to later viral assembly stages and release. SUMOylation of HIV-1 p6 (gag proteolytic product) has a negative impact during early viral replication [52]. SUMOylation limits another HIV-1 protein, integrase (IN). Mutations in the phylogenetically conserved SCMs (SUMOylation consensus motifs) of HIV-1 IN protein results in the reduction of infectivity, replication kinetics, and integration events [53]. In addition to SUMOylating the viral proteins, host proteins belonging to pathways such as cell cycle, mRNA processing, transcription elongation, and immune pathways are often SUMO-modified [54]. The earlier investigation has shown SUMOylation of RIG-1 during RNA virus infection promotes its ubiquitination, leading to increased interaction with Cardiff, a mitochondrial adaptor in RIG-1 anti-viral pathway resulting in enhanced IFN signaling, which affects viral replication (Figure 2) [55].
2.3.2. Role of SUMOylation in the Promotion of Viral Replication
Contrary to the SUMO modifications mentioned earlier in viral proteins, which leads to inhibition of viral replication, SUMOylation modification of proteins also results in enhancing viral replication e.g., ebola virus VP35 protein promotes the SUMOylation of host interferon response components (IRF3 and IRF7) via SUMO E3 ligase PIAS1, significantly hampering IFN promoter activity (Figure 2) [56]. Sumoylation of SARS-CoV NP promotes viral RNP formation and nucleocapsid assembly, which favor viral replication and replication [57]. SUMOylation of IAV matrix protein (M1) and NP proteins results in the efficient assembly of viral RNP-M1 complexes and consequent viral maturation and release (Figure 2) [58][59]. In addition to this, SUMOylation inhibits IFN response in various viruses that promote viral growth, e.g., Ebola virus VP24 protein upon sumoylation binds to USP7 and is deubiquitinated. It binds to karyopherin alpha1, a STAT1 NLS (Nuclear localization signal) receptor, and inhibits IFN response [60][61], thus favoring viral replication. SUMOylated ZIKV NS5 binds to STAT1 and prevents its association with PML (promyelocytic leukemia), which is needed for ISG expression leading to reduced ISG expression [62].
2.4. NEDDylation
NEDDylation is a process where target proteins undergo covalent attachment to ubiquitin-like molecule NEDD8 (neural precursor cell expressed, developmentally down-regulated 8) [63]. NEDDylated substrates have been implicated in cancer, DNA damage responses, transcriptional regulation, nucleolar stress signaling, apoptosis, and ubiquitin-proteasome machinery [9]. NEDDylation, an essential regulator of CRL (Cullin-RING ubiquitin ligase) activation, is an attractive target for viruses to hijack host ubiquitin machinery. By doing so, viruses modulate myriads of cellular pathways enhancing viral survival and growth in the host [64].
Although some viral proteins have also been identified as targets of neddylation, most studies focus on ubiquitin machinery’s viral intervention via neddylation. Modulation in the neddylation pathway has been studied in very few representative viruses such as influenza and HIV [5][65]. For the context of this review, we will focus mainly on the intervention of RNA viruses in host neddylation machinery and vice versa.
2.4.1. Role of NEDDylation in the inhibition of viral replication
NEDDylation, like other ubiquitin-like modifiers, is known to target the viral proteins to degradation (Figure 2) [5]. Upon viral entry into the cell, multiple anti-viral cascades are triggered. One of them involves phosphorylation and homodimerization of IRF3, leading to its activation. Activated IRF-3 is translocated to the nucleus, where it acts as a transcription factor for multiple anti-viral genes, including IFN-I, thus mounting cellular defenses[66]. To counteract the IFN response, viruses cause degradation of IRF-3 by hijacking RING-ligases. For example, the Sendai virus (SeV) upon infection causes neddylation-dependent proteasomal degradation of IRF3 by utilizing Cul1 based ubiquitin ligases [67]. HDM2 E3 ligases target IAV PB2 (Polymerase basic protein 2) for neddylation (K699). Conjugation of NEDD8 to IAV PB2 harm the protein stability and viral replication. In contrast, the mutant (K699R) showed heightened virulence during infection in mice, highlighting the importance of neddylation on viral proteins and replication [5]. IRF3 and IRF7 were also identified as targets for NEDD8 conjugation [49], suggesting a direct link between the neddylation of viral proteins and viral replication inhibition. Moreover, the study established that NEDD8 deficient zebrafish larvae and adults were more susceptible to SVCV (Spring viremia of carp virus) infection than wild type [49].
2.4.2. Role of NEDDylation in the promotion of viral replication
NEDDylation of proteins leads to their degradation, and this has a pro-viral effect when the modification occurs on the immune pathway proteins. During viral infection, this hampers the immune pathway and promotes viral replication. The neddylation of CRL4 and CRL5 causes ubiquitin-mediated proteasomal degradation of APOBEC3G (A3G) (which belongs to the family of cytidine deaminases and restricts retroviral replication to protect the infected host by inducing hypermutations) and SAMHD1 (a deoxynucleoside triphosphohydrolase that blocks retrovirus infection at reverse transcription). It promotes viral replication (Figure 2) [51][68]. Neddylation of Culin-1 during Influenza virus infection cause its inhibition and negatively affect the NF-kB pathway. Inhibition of the neddylation pathway by targeting Nedd8-activating enzyme subunit 1 (NAE1) with inhibitor (MLN4924) suppresses virus replication [69], suggesting the important role of NEDD8 in viral replication.
3. Carbohydrate-based post-translational modifications
Carbohydrate moieties as mono/poly and homo/heteropolymers are attached to the proteins. The transfer of moieties is facilitated by a special class of enzymes called glycosyltransferases [70]. These modifications serve the purpose of solubilization, receptor-ligand binding, which is crucial during viral protein function, viral complex assembly, viral entry, host-pathogen interactions, viral protein secretion, virulence, and antigenicity [71]. Beside eukaryotes, where it is well known to play a role in cells’ function, it also plays a vital role in the life cycle of viruses such as HIV, flavivirus, alphavirus, influenza, and coronavirus [72]. The envelope and secreted viral proteins are modified with carbohydrate moieties, where these moieties assist in protein folding and assembly. Glycosylation of secreted viral proteins often subverts humoral immune response [72][73].
3.1. Glycosylation
Glycoconjugates are macromolecules composed of carbohydrate chains (made up of homo or heteropolymers of either fucose, mannose, sialic acid, N-acetylglucosamine (GlcNAc), or Galactose) covalently attached to the protein in a process termed as glycosylation [74][75]. An array of the glycoconjugates exist within the intracellular and extracellular milieu, and these differ in the glycan composition, anomeric ring linkages, length, and site of occurrence. Glycosylation of proteins is classified into mainly two types: N-linked glycosylation, where the glycan moiety is linked to asparagine, and O-Linked glycosylation, where the glycan moiety is attached to oxygen atom present on the hydroxyl group of serine or threonine amino acid residue within a protein [74]. In addition to playing an essential role in cells' functioning, glycosylation plays a significant role in the life cycle of RNA viruses, including from genus Alphavirus, Flavivirus, Ebolavirus, Betacoronavirus, Arenavirus, and Henipavirus [71]. The glycan moieties play a crucial role in viral entry, assembly, virulence, pathogenicity (Figure 3) and are discussed below.
3.1.1. Role of glycosylation in the inhibition of viral replication
Glycosylation help viruses at various stages of their life cycle. It also favours the host by recognizing viral proteins, activating the immune system. Viral proteins, besides helping in virus replication, assembly are internalized by immune cells (macrophages and dendritic cells) and results in MHC (Major histocompatibility complex) mediated antigen presentation and virus epitope-specific antibody generation [76][77]. The antibodies are glycosylated at Fc (fragment crystallizable) region before their secretion. Glycosylation helps in their solubilization and complement activation. Their glycosylation pattern changes among different viruses such as HIV-1, Influenza, DENV, and Ebola and has different avidity and affinity [78]. Glycosylation affects humoral response via the complement system recruitment [79], which leads to restriction in viral replication. Secreted antibodies play a role in restricting viral infection by virus neutralization, complement activation, and antibody-dependent cellular phagocytosis (ADCP). Antibodies against viral proteins such as HCV E2 protein restrict viral infection by binding to viral proteins involved in assembly or pathogenicity [80], thereby restricting viral replication. Antibody binding to viruses/viral proteins leads to the formation of complexes that are recognized by immune cells leading to their phagocytosis mediated clearance. This phenomenon is observed in HIV-1, influenza; however, there are few exceptions like ZIKV, DENV, yellow fever virus where this leads to enhancement in viral replication due to antibody-dependent enhancement (ADE) [81][82][83], a phenomenon of phagocytosis of virus antibody complex which has higher efficiency of entry but escapes phagocytic degradation due to non-neutralizing antibodies which lead to viral escape. In addition to antibodies, Mannose-binding lectin (MBL) also restricts viral infection. MBLs are pattern binding proteins that differentiate self from non-self protein based on their glycosylation pattern with their carbohydrate recognition domains (CRDs). These are known to inhibit viral (SARS-CoV and DENV) replication by activating a lectin-based complement system (Figure 3) [84][85].
3.1.2. Role of glycosylation in the promotion of viral replication
Viruses being opportunistic pathogens, depends on the host glycosylation machinery for glycan conjugation on their proteins [73]. Glycosylation is involved at various stages of viral replication to promote its amplification. These involve (i) Receptor binding of the virus inside the cell. Glycosylation assists viruses in receptor binding and subsequent infection of immune cells. DC-SIGN (Dendritic cells-soluble extracellular dendritic cell-specific ICAM3 grabbing non-integrin) receptors present on the surface of immune cells such as dendritic cells and macrophages facilitate viruses such as alphavirus (SINV) and flavivirus (DENV) entry by interacting with glycans frequently present on the viral envelope proteins (Figure 3) [86][87]. The Ebola virus attachment and entry into cells is mediated by surface GP1 (glycoprotein subunit 1). The GP1 subunit mediates receptor binding and is enriched by O-linked glycan moieties [88]. The glycan moieties shield antigenic epitopes on the GP1 itself and impair antigenic presentation by sterically hindering accessibility to host surface proteins such as MHV-I and integrins, thus severely impending host immune response (Figure 3) [89]. Like Ebola, viral attachment glycoprotein (G) encoded by the RSV (Respiratory syncytial virus) has high glycosylated O-glycans and has a role in the fusion of virus and cell membrane [90].
(ii) Viral replication and maturation. Glycosylation of viral envelop proteins is the best-known example of glycosylation in the virus. The modification helps the proteins to adopt confirmation and ultimately help in replication as well as maturation (Figure 3). Few examples are: envelop protein of West Nile virus (WNV), Influenza, HIV-1, and ZIKV [91][92][93]. The importance of the glycosylation on envelope proteins can be understood from the fact that inhibition of glycosylation severely affects the maturation process in viruses such as Ebola, Influenza, HIV-1, and ZIKV [92][93].
(iii) Viral pathology. Glycans also play a crucial role in pathology during viral infection. Glycosylation promotes pathogenesis by enhancing the binding of viral proteins to cells leading to enhanced infection (Figure 3). N glycan moieties on envelop protein in ZIKV, Influenza, HIV-1, Ross River virus (RRV) promote virulence [93][94][95]. In addition to glycosylation of structural proteins, non-structural proteins such as DENV NS1 are glycosylated and have a role in protein secretion and stability of hexameric form which helps in immune evasion by binding to lectin pathway proteins such as C1s, C4, C4-binding protein, MBL and prevents lectin complement activation and DENV neutralization, thus regulate pathogenesis [96][97].
(iv) Immune evasion. Glycosylation helps the virus to escape the immune system by avoiding recognition. Some viruses display heavily glycosylated surface envelope proteins to escape humoral immune recognition. HIV-1 envelope is a trimer of non-covalent gp120-gp41 dimers. These subunits contain numerous glycan moieties with a gp120 monomer alone constituting 18–33 glycans (oligo-mannose type) and act as a shield against the immune system [98]. The glycan moieties are weakly immunogenic by themselves, and the glycan shield sterically hinders neutralizing antibodies from penetrating and binding to epitopes on the underlying envelope protein (Figure 3) [99]. However, glycan holes, which represent areas close to the inter-protomer axis, the CD4-binding site, and the fusion peptide, are accessible to neutralizing antibodies [100].
3.2. ADP-Ribosylation
ADP-ribosylation is a ubiquitous modification present across all life domains ranging from the virus to eukaryotic organisms [101][102][103]. ADP-ribosylation utilizes nicotinamide adenine dinucleotide (NAD) as a cofactor to transfer ADP-ribose nucleotide onto proteins and DNA [101]. The reaction is catalyzed by the enzyme ADP-ribosyl transferases known as ARTDs (ADP-ribosyltransferase, diphtheria toxin-like) or PARPs (Poly ADP ribose polymerase) [101]. The ADP-ribosylation plays an essential role in multiple biological processes such as viral replication, translation, signal transduction, epigenetics, cellular stress response, and protein degradation [104]. Modification by PARPs occurs mostly on acidic amino acid residues such as glutamate and aspartate, but other residues such as serine, arginine, lysine, and cysteines can also act as acceptors [102]. The role of ADP-ribosylation in promoting and inhibiting viral replication is mentioned in the section below and Figure 3.
3.2.1. Role of ADP Ribosylation in the Inhibition of Viral Replication
PARPs regulate innate immune response at various steps of viral infection. ADP-ribosylation restricts/inhibit viral infection by (i) induction of IFN response. Hosts use MARylating (Mono ADP ribosylating) and non-enzymatic PARPs to modulate IFN response and pro-inflammatory cytokine induction. PARP binds to the RIG-1 receptor and promotes its oligomerization and initialization of the signaling cascade [105]. Using a Venezuelan equine encephalitis virus (VEEV) mutant-based model, Atasheva et al. (2012) identified PARP12, among other PARP proteins, as an essential ISG involved in cellular defense against numerous alphavirus [106].
(ii) Transcriptional silencing of retroviral genome. PARP1 leads to transcriptional silencing of the integrated HIV genome in host cell either by integrating the HIV genome in transcriptionally disfavored regions such as the centromere region of chromosomes (Figure 3) or by epigenetic mechanisms, thereby repressing viral infection [107][108]. (iii) Proteasomal degradation of viral proteins and RNA. ZIKV encoded NS1 and NS3 proteins undergo PARP12-mediated PARylation (poly ADP ribosylation). The PAR modification further recruits E3 ubiquitin ligase for ubiquitination. Thus, these two modifications work in synchrony to bring about proteasomal degradation of ZIKV proteins (NS1 and NS3) (Figure 3) [109]. PARP13 is one of the first PARP enzymes for which anti-viral function was identified, and two isoforms ZAP-S (N-terminal tandem zinc-finger motifs that bind RNA) and ZAP-L (N-terminal tandem zinc-finger with C-terminal PARP inactive catalytic domain). PARP13/ZAP-S via N-terminal domain inhibits viral replication by promoting viral RNA degradation by recruiting RNA processing exosomes in HIV-1, Semiliki forest virus [110][111], SINV, and Moloney murine leukemia virus (MLV), alternatively it also binds to 3’-UTR of IFN and causes degradation of IFN (IFNL1, IFNL2, and IFNβ) mRNA [112]. Other PARPs known to inhibit viral replication include PARP5a, PARP7, PARP9, PARP10, PARP12, and PARP13, which inhibit viral replication in the ways such as increasing IFNs, ISGs, binding to viral RNA, and preventing its translation [105].
3.2.2. Role of ADP Ribosylation in Promotion of Viral Replication
ADP ribosylation, in addition to inhibiting viral replication, also promotes viral replication. It does so by (i) Inhibition of the IFN response. During IAV replication, PARP1 is localized to the cytosol, where it mediates the IFN alpha receptor (IFNAR) degradation resulting in impaired host anti-viral defense. PARP1 mediated IFNAR degradation depends on PAR-enzymatic activity, although the mechanism is still unclear (Figure 3) [65]. Thus, ADP ribosylation has a potential pro-viral role during influenza virus infection. PARP11 induced during VSV infection causes mono ADP ribosylation of ubiquitin E3 ligase β-transducin repeat-containing protein (β-TrCP), which leads to ubiquitination of IFNAR1 and its degradation. This results in reduced IFN and anti-viral response [113]. To counter host PARPs mediated immune response, several RNA viruses such as alphaviruses, hepatitis E virus, coronavirus [114][115][116] has macrodomain and possess the ability to recognize and reverse the effect of ADP ribosylation. Moreover, the SARS-CoV macrodomain mutant induces a robust pro-inflammatory cytokine response and increases the virus sensitivity to the IFN-1 treatment [116].
(ii) Induction of viral replication. PARP1 is activated upon double-stranded breaks and modifies nuclear histones leading to chromatin decondensation and enhanced access to repair enzymes. The enzyme facilitates HIV-1 integration within the host genome by mediating DNA repair events that follow viral integration (Figure 3) [117]. Macrodomain from the RNA virus families bind both mono and poly-ADP-ribose and can hydrolyze and remove mono-ADP-ribose from proteins [118] and promote the replication of viruses [116][119]. CHIKV nsP3 protein macrodomain mutants showed that ADP-ribose binding caused the viral replication initiation, while hydrolase activity is essential in amplifying replication complexes [119]. One other study showed the importance of macrodomain in the HEV (Hepatitis E virus) replication where it was found that macrodomain mutants (N809A and H812L) were nonviable, whereas RNA replication was abrogated in mutants (G816A and G817A) [115]. Another study by Li et al. showed a strong correlation between viral replication and enzymatic activity of the mutant [120]. These findings suggest that macrodomain is not only crucial in replication but also post RNA replication.
4. Lipidation
Lipids are significant components of cell membranes and help to maintain cell homeostasis. In addition to their role in the membrane, these are also directly attached to the proteins in the process called protein lipidation. Protein lipidation is the covalent attachment of lipid moieties to proteins [121]. It is an essential post-translation modification involved in processes like membrane localization of proteins, protein-protein interactions, protein stability, enzymatic activity [122][123]. Some of the standard types of lipidation examples are palmitoylation, prenylation, and myristoylation [121].
4.1. Palmitoylation
Palmitoylation is a reversible type of enzymatic modification in which palmitoyl chain is attached to a cysteine residue called S-palmitoylation involving attachment of 16-carbon palmitoyl group via a thioester linkage) [121]. S-palmitoylation attachment regulates the functions of the proteins by regulating their association with membranes, compartments, trafficking, cellular localization of proteins, and stability [124]. The enzyme responsible for the modification is called Palmitoyltransferase, such as GODZ (Golgi associated DHHC (Asp-His-His-Cys) zinc finger domain, also known as such as DHHC3 [125]. Initial reports of palmitoylation of envelope glycoproteins of viruses were in SINV and VSV [126][127].
4.1.1. Role of the Palmitoylation in the Inhibition of Viral Replication
The palmitoylation of host proteins plays a vital role in the anti-viral defense strategy. TLRs (Toll-like receptors) recognize nucleic acids of the viruses and triggers the production of type I interferons (IFNs), mainly INFα and IFNβ, and induce an anti-viral reaction in an autocrine and paracrine manner [128]. Interferon-induced transmembrane proteins (IFITMs) are potential viral restriction factors against a wide range of enveloped viruses such as IAV, WNV, DENV, and ZIKV. IFITMs are primarily located in endolysosomal membranes and facilitate the virus’ degradation by blocking their fusion with the membrane (Figure 3) [129][130]. It has also been found that the S-palmitoylation also facilitates the clustering of IFITM3 in the membranes, which suggests that it shows a potential role for its anti-viral activity [131]. Hence, in this way, various host proteins play a crucial role in affecting both the virulence and the host’s immune reactions.
4.1.2. Role of the Palmitoylation in the Promotion of Viral Replication
During the infection, palmitoylation contributes to the virus growth by enhancing virulence, infectivity, cell fusion, viral protein localization, and virion assembly [132][133][134][135]. Palmitoylated viral proteins can be categorized into three categories a) spike proteins, b) viroporins, and c) peripheral membrane protein [136]. Palmitoylation of HCV NS2 is required for NS2-NS3 autoprocessing and viral assembly, thus, this has an important role in RNA replication (Figure 3) [137]. Similarly, HEV ORF3 is also palmitoylated and plays a role in viral secretion [138]. HIV-1 glycoprotein palmitoylation is required for gp160 incorporation and infectivity [133]. Alphavirus TF (transframe) protein is palmitoylated and is involved in its localization to the plasma membrane and incorporation into a virus [134], and palmitoylated nsP1 is involved in anchoring replication complex to the plasma membrane [139]. The examples mentioned above show that the palmitoylation of viral proteins is crucial for viral replication.
4.2. Myristoylation
Myristoylation is the attachment of the myristoyl group (a 14-carbon fatty acid) by N-myristoyltransferases (NMT) to the N-terminal glycine of host and viral proteins. The modification involves protein targeting to membranes, protein-protein interactions, and virus entry [140]. Viral proteins of several viruses such as HIV-1, picornaviruses, Lassa virus, DENV [140][141][142] are known to myristoylated and play an important role in the viral cycle. Inhibition of myristoylation by chemicals such as DDD85646, a pyrazole, and NMT inhibitor, or its knockdown significantly reduces the virus yield indicating an important role of myristoylation in virus maturation and infectivity [143][144].
4.2.1. Role of Myristoylation in the Promotion of Viral Replication
Myristoylation of viral proteins like other PTMs helps the viral life cycle processes. HIV-1 matrix protein 31 (MA31) and Nef proteins are myristoylated and have a role in virus assembly and replication (Figure 3). MA31 helps the viral assembly at membrane and myristoylation of MA31 enhances the affinity of protein to the membrane, thus helps in assembly. Nef is an early infection protein that increases pathogenicity by downregulating CD4 expression in T cells and helping in virion release [145]. Myristoylation of matrix protein Z of Lassa virus, VP4 protein of enterovirus 71 (EV71), and poliovirus polyproteins help in membrane localization and virus assembly, thus in virus replication [141][146][147].
4.3. Prenylation
Prenylation is essential for many cellular processes, including signal transduction, cytoskeletal reorganization, and membrane trafficking [163]. It involves the attachment of isoprene lipids (15-carbon (farnesyl) or a 20-carbon (geranylgeranyl)) to the cysteine residue of the proteins. Proteins have CAAX residues at C-terminal (where “C” is cysteine, “A” generally represents an aliphatic amino acid, and the “X residue is serine, methionine, or glutamine) [148]. There are few reports of the role of prenylation during viral infection. In silico analysis predicted prenylation of viral proteins of HIV-1, IAV, HCV, but they are yet to be confirmed.
4.3.1. Role of Prenylation in the Inhibition of Viral Replication
Long isoform of ZAP (Zinc finger anti-viral protein) protein is farnesylated and is targeted to the membrane. The protein inhibits replicating viruses of the Togaviridae family such as SINV, Semliki virus (SFV), and RRV by binding to incoming viral RNA (Figure 3) [149].
4.3.2. Role of Prenylation in the Promotion of Viral Replication
FBL2 (F-box and leucine-rich repeat protein 2), a component of the E3 ubiquitin ligase complex protein, is geranylgeranylated, which interacts with NS5A and plays a role in HCV replication [150]. In addition to host proteins, proteins from several viruses are farnesylated, including the hepatitis delta virus large antigen (lHDAg) [151]. The protein is prenylated at the terminal CXXX box and helps in viral assembly [152]. During IAV infection, geranylgeranylation of mitochondrial anti-viral signaling protein (MAVS) inhibits their anti-viral signaling by translocation of Rac1, which is a guanosine triphosphatase to mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs). Rac1 recruitment to MAVS signalosome inhibits the interaction of TRIM31 with MAVS. Rac1 also facilitates the recruitment of Caspase-8, which cleaves MAVS and inhibits MAVS-mediated interferon signaling by cleaving ubiquitinated Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) [153].
5. Small Chemical Groups based on Post-Translational Modifications
Small chemical groups such as phosphate (PO43-), methyl (-CH3), acetyl (-COCH3) from the side chain of amino acids of proteins. These modifications are catalyzed by specific enzymes belonging to families such as kinases/phosphatase, methyltransferase/demethylase, and acetyltransferases/deacetylase. These modifications generally alter the surface charge or act as a binding site for interaction with other proteins [154]. These are well known to play roles in cellular pathways such as replication, metabolic pathways, signaling pathways, and immune pathways [155][156][157][158].
5.1. Phosphorylation
Phosphorylation is one of the most conserved types of PTM. The phosphate group is added to amino acids such as serine, threonine (Thr), or tyrosine (Tyr) residues on proteins. It requires adenosine triphosphate (ATP) and is a reversible reaction involving protein kinases and protein phosphatases [155][159]. The phosphorylation of viral and cellular proteins can significantly impact viral infection, replication, and cytotoxicity in a host cell. Eukaryotic cells use these modifications to control the functional repertoire of proteins. Most viral proteins mimic key regulatory factors to usurp this host machinery and promote efficient viral outcomes. Phosphorylation plays a vital role in protein: protein interactions, protein stability, signal transduction, transcription regulation, intracellular localization, cell cycle progression, and apoptosis [160]. Protein phosphorylation is also essential for many intracellular obligate pathogens to establish a productive infection cycle.
5.1.1. Role of Phosphorylation in the Inhibition of Viral Replication
RNA viruses such as VSV, SINV, SeV, and IAV induce interferon response, restricting viral infection [161]. During RNA viral infection, the dsRNAs are recognized by pattern recognition molecules such as RIG-I and dsDNA made during retroviral replication bind cGAS and are converted to cyclic dinucleotides. Both these lead to activation of STING and its translocation to perinuclear vesicles. This leads to the induction of chemokines, type 1 interferon genes, and pro-inflammatory genes via the NF-kB pathway and involves TBK1 (TANK-binding kinase 1) phosphorylation, which in turn phosphorylate IRF3 and STAT6. The cytokines bind to receptors in the neighboring cells, leading to phosphorylation mediated dimerization of STAT protein, which are then translocated to the nucleus and activate the JAK-STAT pathway (Figure 4) [162][163]. Influenza NS1 is the canonical antagonist of innate immune responses. It binds to viral RNA, TRIM25, RIG-1, and promotes viral replication and reduces interferon response, but upon phosphorylation during the later stage of infection, its binding to viral RNA, TRIM25, and RIG-1 reduces, thereby negatively affecting viral replication and immune response [164]. Phosphorylation of IAV NS1 and Rubella capsid disrupts protein–RNA interactions and protein-protein interactions (Figure 4) [157][165], thereby affecting the viral replication and assembly.
5.1.2. Role of Phosphorylation in the Promotion of Viral Replication
Phosphorylation is involved in signaling pathways and promotes viral growth by (i) increasing the viral replication. Alphaviral nsP3 is heavily phosphorylated at the C-terminal hypervariable domain (HVD), and it shows significant variability in sequence among alphaviruses [166]. In the Semliki Forest virus (SFV), nsP3 C-terminal tail is involved in the activation of the phosphatidylinositol-3-kinase (PI3K)-Akt-mammalian target of Rapamycin (mTOR) pathway as well as replication complex formation. The hyperphosphorylated tail of SFV leads to AKT activation, resulting in efficient internalization of replication complexes in SFV, but not the Chikungunya virus (CHIKV) (Figure 4) [167]. Other examples include Ebola VP35, a viral RNA-dependent RNA polymerase cofactor, and its activity is phosphorylation-dependent; thus, phosphorylation has a positive role in Ebola viral replication [168].
(ii) Increasing the viral budding. The phosphorylation of matrix protein Z of the Lassa virus at Y97and S98 of PPXY late domain (Pro-Pro-X-Tyr domain, a motif involved in virus release) regulates the virus release by regulating the budding process. The proteins associated with membrane and help` in the recruitment of NP to the membrane where viral assembly and release takes place [169]. (iii) Inhibition of immune pathways. Flaviviral non-structural protein 5 (NS5) inhibits interferon-mediated anti-viral response by targeting host JAK-STAT protein, inhibition of anti-viral signaling pathways. DENV and other flaviviral such as ZIKV, and Yellow fever virus NS5 bind to STAT2, preventing its phosphorylation (Figure 4), which is required to form a complex with JAK and further translocated to the nucleus for anti-viral responsive gene expression [170] and target it to proteasomal degradation [171][172]. Nucleocapsid (N) protein binds to B23 (a nucleolar phosphoprotein and substrate for CDK2/cyclin E, involved in centrosome replication and regulates cell cycle), STAT, and prevents their phosphorylation. This negatively affects the cell cycle and interferon response [173][174]. SARS-CoV nucleoprotein (N) phosphorylation may also affect its nucleocytoplasmic shuttling by interaction with the host adapter protein [175].
5.2. Methylation
Methylation of viral RNA is a well-studied viral growth regulation mechanism [176], whereas the post-translational role of methylation during RNA viral infection is not well studied. The virus utilizes several approaches for its full replication and immune evasion strategy by co-opting the transcriptional and translational regulation of infected cells to produce new viral particles [177]. S-adenosyl methionine (SAM) acts as a methyl group donor, and transfer is catalyzed by methyltransferases.
5.2.1. Role of Methylation in the Inhibition of Viral Growth
Retroviruses such as HIV integrate into the host genome and be in the latent phase by regulating host histone methylation. SEC (Super Elongation Complex) promotes H3K27 acetylation at HIV-1 LTR (long terminal repeat), which leads to transcription elongation, and the same complex also recruits CARM1, a methyltransferase, and causes H3R26 (histone 3 arginine 26) methylation leading to attenuation [178]. HIV Tat protein promotes the expression of MeCP2 (methyl CpG binding protein 2) and Ezh2 (Enhancer of zeste homolog 2, a histone-lysine N-methyltransferase enzyme), which induces Tri-methylation of H3K27 leading to attenuation [179]. In addition to histones, viral proteins are also methylated, which controls virus latency. Methylation of Tat by SETDB1 inhibits transactivation of HIV LTR [180], whereas demethylation by lysine-specific demethylase 1 (LSD1/KDM1) activates HIV transcription [181]. During influenza infection, LSD1 is induced by IFNα and demethylate and activates IFITM3 leading to infection suppression (Figure 4) [182].
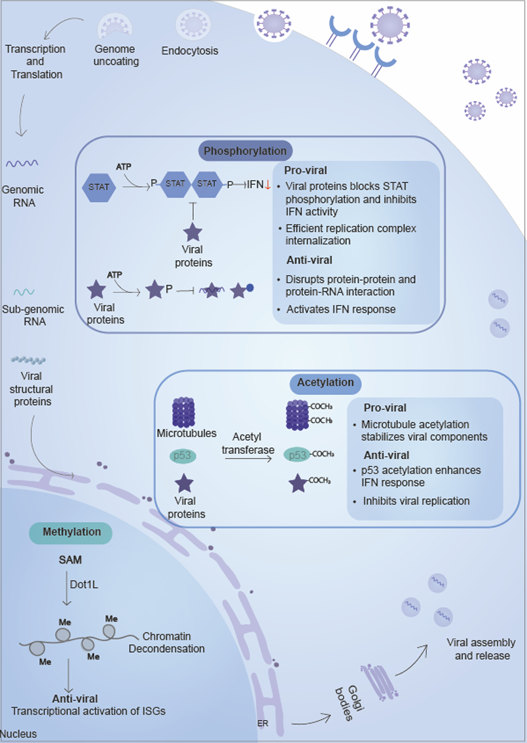
Figure 4. Small molecules/chemical group based PTMs. Small chemical groups such as phosphate, methyl, acetyl, and ionic groups are added to proteins by specific enzymes. Phosphorylation involves kinases which transfer phosphate group and play a role in protein-protein interaction, thereby affecting protein localization and signaling pathways. Methylation of host and viral proteins involved methyl group transfer from substrate protein with the help of methyltransferases. Methylation plays a role in modulating gene expression by regulating chromatin condensation, regulating both host gene expression and transcription of integrated viral genomes. Acetylation of histone proteins regulate gene expression and microtubule organization and affect viral replication.
5.3. Acetylation
Acetylation is one of the most common PTMs present in histone protein. It is involved in the global regulation of gene expression during cell differentiation and development by histone protein modification [183]. Acetylation also affects viruses at different stages of cycles like entry, replication, fusion, transport, and release [184]. The modification is most prevalent on lysine amino acid and is catalyzed by acetyltransferases belonging to the family of GNAT (Gcn5-related N-acetyltransferases), MYST (histone acetyltransferases), and p300/CBP (E1A binding protein p300/CREB-binding protein) localized in the nucleus, cytoplasm as well as in mitochondria [183].
5.3.1. Role of Acetylation in the Inhibition of Viral Replication
Acetylation and deacetylation of viral proteins and human immune pathway proteins affect their activity leading to inhibition of viral replication, e.g., acetyltransferases (PCAF and GCN5) mediated acetylation of influenza nucleoprotein (NP) protein near to RNA binding groove (K31 and K90) inhibit the viral replication (Figure 4) [185]. In addition to directly acetylating/deacetylating viral proteins, the host also modifies its proteins to enhance the anti-viral effect as seen in the p53 protein, which is acetylated during infection and leads to transactivation of pro-apoptotic and IFN-stimulated genes (Figure 4) [186]. Deacetylases are essential in regulating gene expression due to their role in chromatin remodeling. Histone deacetylase (HDAC) removes the acetyl group from histone molecules and inhibit gene expression. NF-kB p50 mediated recruitment of HDAC1 to HIV LTR leads to repression of transcription due to impairment of RNA Pol II recruitment [187]. Other HDAC, HDAC6, causes deacetylation of RIG-I at C-terminal enhances RNA sensing, or prevents the trafficking of IAV virus to membrane or RNA Pol subunit PA leading to proteasome degradation [184][188][189], thereby inhibiting the viral replication.
5.3.2. Role of Acetylation in the Promotion of Viral Replication
Acetylation of influenza viral protein such as NP (K77, K113, and K229) and NS1 (K108) play a positive role in their replication and growth via increase the protein’s activity and IFN antagonism [190][191][. Acetylation of viral proteins such as HIV Tat promotes transcription of the integrated genome by recruiting the positive transcription elongation factor (pTEFb) complex at LTR promoter [181]. In contrast to inhibiting viral replication, HDAC6, in association with rabies matrix protein (M), promotes viral replication by depolymerizing tubulin [192]. Another HDAC, HDAC4 during Sendai virus infection, binds to TBK1/IKKε and prevents phosphorylation of IRF3, which is needed for the production of type I IFN [193]. Microtubules are structural components and are involved in providing structural support as well as cellular transport. Viral infections are found to induce acetylation of microtubules and stabilize viral compartments in the cytoplasm of cells in the case of noroviruses, rotavirus, and reovirus, and promotes viral replication (Figure 4) [194][195]. HDAC inhibition using chemical inhibitors in some viruses such as Japanese encephalitis virus (JEV) and RSV results in the inhibition of viruses, indicating the role of acetylation in viral replication [196][197].
6. Conclusion
Post-translational modifications of proteins are vital to the cellular processes of both hosts as well as viral proteins. These help to maintain homeostasis by regulating their solubility, stability, interaction with partners, and degradation. The PTMs help the host combat viral infection by activating the immune system and degrading viral proteins and playing a positive role for viruses where they help in viral assembly, the enzymatic activity of the viral protein, and interferon response inhibition. The dominance of the immune system’s activation to control viral replication and control over the immune system decides the outcome of the disease. Recently a few studies have shown that targeting host proteins involved in post-translational modifications such as kinases can be effective in restricting viral replication. Additionally, targeting the proteins involved in the immune response reduces the complications related to immune activation in viral infection such as SARS-CoV-2, IAV, alphavirus, and flavivirus, and could be a way to reduce the damage caused by the host to itself. More future studies about the mechanism and components involved in post-translational modifications in various viruses can reveal more targets for the treatment of infections.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010323
References
- Alexander E. Gorbalenya; Genomics and Evolution of the Nidovirales. Nidoviruses 2014, 1, 15-28, 10.1128/9781555815790.ch2.
- Alexander E. Gorbalenya; Luis Enjuanes; John Ziebuhr; Eric J. Snijder; Nidovirales: Evolving the largest RNA virus genome. Virus Research 2006, 117, 17-37, 10.1016/j.virusres.2006.01.017.
- Ana L. Santos; Ariel B. Lindner; Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxidative Medicine and Cellular Longevity 2017, 2017, 1-19, 10.1155/2017/5716409.
- Anna Rahnefeld; Karin Klingel; Anett Schuermann; Nicola L. Diny; Nadine Althof; Anika Lindner; Philipp Bleienheuft; Konstantinos Savvatis; Dorota Respondek; Elisa Opitz; et al. Ubiquitin-Like Protein ISG15 (Interferon-Stimulated Gene of 15 kDa) in Host Defense Against Heart Failure in a Mouse Model of Virus-Induced Cardiomyopathy. Circulation 2014, 130, 1589-1600, 10.1161/circulationaha.114.009847.
- Tinghong Zhang; Zhen Ye; Xiaohai Yang; Yujie Qin; Yi Hu; Xiaomei Tong; Wenbin Lai; Xin Ye; NEDDylation of PB2 Reduces Its Stability and Blocks the Replication of Influenza A Virus. Scientific Reports 2017, 7, 43691, 10.1038/srep43691.
- Preeti Bharaj; Colm Atkins; Priya Luthra; Maria Isabel Giraldo; Brian E. Dawes; Lisa Miorin; Jeffrey R. Johnson; Nevan J. Krogan; Christopher F. Basler; Alexander N. Freiberg; et al. The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. Journal of Virology 2017, 91, e00833-17, 10.1128/jvi.00833-17.
- Helena Ryšlavá; Veronika Doubnerová; Daniel Kavan; Ondřej Vaněk; Effect of posttranslational modifications on enzyme function and assembly. Journal of Proteomics 2013, 92, 80-109, 10.1016/j.jprot.2013.03.025.
- Kirby N Swatek; David Komander; Ubiquitin modifications. Cell Research 2016, 26, 399-422, 10.1038/cr.2016.39.
- Annemarthe G. Van Der Veen; Hidde L. Ploegh; Ubiquitin-Like Proteins. Annual Review of Biochemistry 2012, 81, 323-357, 10.1146/annurev-biochem-093010-153308.
- Doris Popovic; Domagoj Vucic; Ivan Dikic; Ubiquitination in disease pathogenesis and treatment. Nature Medicine 2014, 20, 1242-1253, 10.1038/nm.3739.
- Jonathan M. D. Vosper; Gary S. McDowell; Christopher J. Hindley; Christelle S. Fiore-Heriche; Romana Kucerova; Ian Horan; Anna Philpott; Ubiquitylation on Canonical and Non-canonical Sites Targets the Transcription Factor Neurogenin for Ubiquitin-mediated Proteolysis. Journal of Biological Chemistry 2009, 284, 15458-15468, 10.1074/jbc.m809366200.
- Cecile M. Pickart; Michael J. Eddins; Ubiquitin: structures, functions, mechanisms. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2004, 1695, 55-72, 10.1016/j.bbamcr.2004.09.019.
- Kezhen Wang; Chunling Zou; Xiujuan Wang; Chenxiao Huang; Tingting Feng; Wen Pan; Qihan Wu; Penghua Wang; Jianfeng Dai; Interferon-stimulated TRIM69 interrupts dengue virus replication by ubiquitinating viral nonstructural protein 3. PLOS Pathogens 2018, 14, e1007287, 10.1371/journal.ppat.1007287.
- Adam J Fletcher; Devin E Christensen; Chad Nelson; Choon Ping Tan; Torsten Schaller; Paul J. Lehner; Wesley I Sundquist; Greg J Towers; TRIM 5α requires Ube2W to anchor Lys63‐linked ubiquitin chains and restrict reverse transcription. The EMBO Journal 2015, 34, 2078-2095, 10.15252/embj.201490361.
- Abhilash I. Chiramel; Nicholas R. Meyerson; Kristin L. McNally; Rebecca M. Broeckel; Vanessa R. Montoya; Omayra Méndez-Solís; Shelly J. Robertson; Gail L. Sturdevant; Kirk J. Lubick; Vinod Nair; et al. TRIM5α Restricts Flavivirus Replication by Targeting the Viral Protease for Proteasomal Degradation. Cell Reports 2019, 27, 3269-3283.e6, 10.1016/j.celrep.2019.05.040.
- Qingchen Zhu; Tao Yu; Shucheng Gan; Yan Wang; Yifei Pei; Qifan Zhao; Siyu Pei; Shumeng Hao; Jia Yuan; Jing Xu; et al. TRIM24 facilitates antiviral immunity through mediating K63-linked TRAF3 ubiquitination. Journal of Experimental Medicine 2020, 217, 6, 10.1084/jem.20192083.
- Hashim Ali; Miguel Mano; Luca Braga; Asma Naseem; Bruna Marini; Diem My Vu; Chiara Collesi; Germana Meroni; Marina Lusic; Mauro Giacca; et al. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nature Communications 2019, 10, 1-15, 10.1038/s41467-019-08810-0.
- Girish Patil; Mengmeng Zhao; Kun Song; Wenzhuo Hao; Daniel Bouchereau; Lingyan Wang; Shitao Li; TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Influenza A Virus Infection. Journal of Virology 2018, 92, 10, 10.1128/jvi.00905-18.
- Masayuki Shirakura; Kyoko Murakami; Tohru Ichimura; Ryosuke Suzuki; Tetsu Shimoji; Kouichirou Fukuda; Katsutoshi Abe; Shigeko Sato; Masayoshi Fukasawa; Yoshio Yamakawa; et al. E6AP Ubiquitin Ligase Mediates Ubiquitylation and Degradation of Hepatitis C Virus Core Protein. Journal of Virology 2006, 81, 1174-1185, 10.1128/jvi.01684-06.
- Sebastian Aguirre; Priya Luthra; Maria T. Sanchez-Aparicio; Ana M. Maestre; Jenish Patel; FrancisE Lamothe; Anthony C. Fredericks; Shashank Tripathi; Tongtong Zhu; Jessica Pintado-Silva; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nature Microbiology 2017, 2, 1-11, 10.1038/nmicrobiol.2017.37.
- Sebastian Aguirre; Ana M. Maestre; Sarah Pagni; Jenish R. Patel; Timothy Savage; Delia Gutman; Kevin Maringer; Dabeiba Bernal-Rubio; Reed S. Shabman; Viviana Simon; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLOS Pathogens 2012, 8, e1002934, 10.1371/journal.ppat.1002934.
- Girish Patil; Lingling Xu; Yakun Wu; Kun Song; Wenzhuo Hao; Fang Hua; Lingyan Wang; Shitao Li; TRIM41-Mediated Ubiquitination of Nucleoprotein Limits Vesicular Stomatitis Virus Infection. Viruses 2020, 12, 131, 10.3390/v12020131.
- Maria I. Giraldo; Oscar Vargas-Cuartas; Juan Carlos Gallego-Gomez; Pei-Yong Shi; Leonardo Padilla; Jhon Carlos Castaño-Osorio; Ricardo Rajsbaum; K48-linked polyubiquitination of dengue virus NS1 protein inhibits its interaction with the viral partner NS4B. Virus Research 2018, 246, 1-11, 10.1016/j.virusres.2017.12.013.
- Yong Ran; Jing Zhang; Li-Li Liu; Zhao-Yi Pan; Yuefeng Nie; Hong-Yan Zhang; Yan-Yi Wang; Autoubiquitination of TRIM26 links TBK1 to NEMO in RLR-mediated innate antiviral immune response. Journal of Molecular Cell Biology 2015, 8, 31-43, 10.1093/jmcb/mjv068.
- Marwah Karim; Elise Biquand; Marion Declercq; Yves Jacob; Sylvie Van Der Werf; Caroline Demeret; Nonproteolytic K29-Linked Ubiquitination of the PB2 Replication Protein of Influenza A Viruses by Proviral Cullin 4-Based E3 Ligases. mBio 2020, 11, 8, 10.1128/mbio.00305-20.
- Yu-Chen Lin; King-Song Jeng; Michael Ming-Chiao Lai; CNOT4-Mediated Ubiquitination of Influenza A Virus Nucleoprotein Promotes Viral RNA Replication. mBio 2017, 8, e00597-17, 10.1128/mbio.00597-17.
- James Kirui; Arindam Mondal; Andrew Mehle; Ubiquitination Upregulates Influenza Virus Polymerase Function. Journal of Virology 2016, 90, 10906-10914, 10.1128/jvi.01829-16.
- Sathish Kumar; Rina Barouch-Bentov; Fei Xiao; Stanford Schor; Szuyuan Pu; Elise Biquand; Albert Lu; Brett D. Lindenbach; Yves Jacob; Caroline Demeret; et al. MARCH8 Ubiquitinates the Hepatitis C Virus Nonstructural 2 Protein and Mediates Viral Envelopment. Cell Reports 2019, 26, 1800-1814.e5, 10.1016/j.celrep.2019.01.075.
- Atsushi Okumura; Paula M. Pitha; Ronald N. Harty; ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proceedings of the National Academy of Sciences 2008, 105, 3974-3979, 10.1073/pnas.0710629105.
- Fumihiko Okumura; Akiko J. Okumura; Keiji Uematsu; Shigetsugu Hatakeyama; Dong-Er Zhang; Takumi Kamura; Activation of Double-stranded RNA-activated Protein Kinase (PKR) by Interferon-stimulated Gene 15 (ISG15) Modification Down-regulates Protein Translation. Journal of Biological Chemistry 2012, 288, 2839-2847, 10.1074/jbc.m112.401851.
- Chen Zhao; Haripriya Sridharan; Ran Chen; Darren P. Baker; Shanshan Wang; Robert M. Krug; Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nature Communications 2016, 7, 12754, 10.1038/ncomms12754.
- Meike Kespohl; Clara Bredow; Karin Klingel; Martin Voß; Anna Paeschke; Martin Zickler; Wolfgang Poller; Ziya Kaya; Johannes Eckstein; Henry Fechner; et al. Protein modification with ISG15 blocks coxsackievirus pathology by antiviral and metabolic reprogramming.. Science Advances 2020, 6, eaay1109, 10.1126/sciadv.aay1109.
- Yang Du; Tianhao Duan; Yanchun Feng; Qingxiang Liu; Meng Lin; Jun Cui; Rong-Fu Wang; LRRC25 inhibits type I IFN signaling by targeting ISG15‐associated RIG‐I for autophagic degradation. The EMBO Journal 2017, 37, 351-366, 10.15252/embj.201796781.
- Matthew B Frieman; Kiira Ratia; Robert E. Johnston; Andrew D. Mesecar; Ralph S. Baric; Severe Acute Respiratory Syndrome Coronavirus Papain-Like Protease Ubiquitin-Like Domain and Catalytic Domain Regulate Antagonism of IRF3 and NF-κB Signaling. Journal of Virology 2009, 83, 6689-6705, 10.1128/jvi.02220-08.
- Annette Flotho; Frauke Melchior; Sumoylation: A Regulatory Protein Modification in Health and Disease. Annual Review of Biochemistry 2013, 82, 357-385, 10.1146/annurev-biochem-061909-093311.
- Van G. Wilson; Introduction to Sumoylation. Neurotransmitter Interactions and Cognitive Function 2017, 963, 1-12, 10.1007/978-3-319-50044-7_1.
- Roger D. Everett; Chris Boutell; Benjamin G. Hale; Interplay between viruses and host sumoylation pathways. Nature Reviews Microbiology 2013, 11, 400-411, 10.1038/nrmicro3015.
- Cagan Gurer; Lionel Berthoux; Jeremy Luban; Covalent Modification of Human Immunodeficiency Virus Type 1 p6 by SUMO-1. Journal of Virology 2005, 79, 910-917, 10.1128/jvi.79.2.910-917.2005.
- Alessia Zamborlini; Audrey Coiffic; Guillaume Beauclair; Olivier Delelis; Joris Paris; Yashuiro Koh; Fabian Magne; Marie-Lou Giron; Joelle Tobaly-Tapiero; Eric Deprez; et al. Impairment of Human Immunodeficiency Virus Type-1 Integrase SUMOylation Correlates with an Early Replication Defect. Journal of Biological Chemistry 2011, 286, 21013-21022, 10.1074/jbc.m110.189274.
- Patricia Domingues; Filip Golebiowski; Michael H. Tatham; Antonio M. Lopes; Aislynn Taggart; Ronald T. Hay; Benjamin G. Hale; Global Reprogramming of Host SUMOylation during Influenza Virus Infection. Cell Reports 2015, 13, 1467-1480, 10.1016/j.celrep.2015.10.001.
- Zhiqiang Mi; Jihuan Fu; Yanbao Xiong; Hong Tang; SUMOylation of RIG-I positively regulates the type I interferon signaling. Protein & Cell 2010, 1, 275-283, 10.1007/s13238-010-0030-1.
- Tsung-Hsien Chang; Toru Kubota; Mayumi Matsuoka; Steven Jones; Steven B. Bradfute; Mike Bray; Keiko Ozato; Ebola Zaire Virus Blocks Type I Interferon Production by Exploiting the Host SUMO Modification Machinery. PLOS Pathogens 2009, 5, e1000493-e1000493, 10.1371/journal.ppat.1000493.
- Zheng Fan; Yue Zhuo; Xinyu Tan; Zhi Zhou; Jiangang Yuan; Boqin Qiang; Jinghua Yan; Xiaozhong Peng; George F. Gao; SARS‐CoV nucleocapsid protein binds to hUbc9, a ubiquitin conjugating enzyme of the sumoylation system. Journal of Medical Virology 2006, 78, 1365-1373, 10.1002/jmv.20707.
- Chung-Yi Wu; King-Song Jeng; Michael M.-C. Lai; The SUMOylation of Matrix Protein M1 Modulates the Assembly and Morphogenesis of Influenza A Virus. Journal of Virology 2011, 85, 6618-6628, 10.1128/jvi.02401-10.
- Qinglin Han; Chong Chang; L. Li; Christoph Klenk; Jinke Cheng; Yixin Chen; Ningshao Xia; Yuelong Shu; Z. Chen; Gülsah Gabriel; et al. Sumoylation of Influenza A Virus Nucleoprotein Is Essential for Intracellular Trafficking and Virus Growth. Journal of Virology 2014, 88, 9379-9390, 10.1128/jvi.00509-14.
- Santiago Vidal; Ahmed El Motiam; Rocío Seoane; Viktorija Preitakaite; Yanis Hichem Bouzaher; Sergio Gómez-Medina; Carmen San Martín; Dolores Rodríguez; María Teresa Rejas; Maite Baz-Martínez; et al. Regulation of the Ebola Virus VP24 Protein by SUMO. Journal of Virology 2019, 94, 6, 10.1128/jvi.01687-19.
- St. Patrick Reid; Lawrence W. Leung; Amy L. Hartman; Osvaldo Martinez; Megan L. Shaw; Caroline Carbonnelle; Viktor E. Volchkov; Stuart T. Nichol; Christopher F. Basler; Ebola Virus VP24 Binds Karyopherin α1 and Blocks STAT1 Nuclear Accumulation. Journal of Virology 2006, 80, 5156-5167, 10.1128/jvi.02349-05.
- Jonas N. Conde; William R. Schutt; Megan Mladinich; Sook-Young Sohn; Patrick Hearing; Erich R. Mackow; NS5 Sumoylation Directs Nuclear Responses That Permit Zika Virus To Persistently Infect Human Brain Microvascular Endothelial Cells8. Journal of Virology 2020, 94, 8, 10.1128/jvi.01086-20.
- Guangqing Yu; Xing Liu; Jinhua Tang; Chenxi Xu; Gang Ouyang; Wuhan Xiao; Neddylation Facilitates the Antiviral Response in Zebrafish. Frontiers in Immunology 2019, 10, 1432, 10.3389/fimmu.2019.01432.
- Radoslav I Enchev; Brenda A. Schulman; Peter Matthias; Protein neddylation: beyond cullin–RING ligases. Nature Reviews Molecular Cell Biology 2014, 16, 30-44, 10.1038/nrm3919.
- David J. Stanley; Koen Bartholomeeusen; David C. Crosby; Dong-Young Kim; Eunju Kwon; Linda Yen; Nathalie Caretta Cartozo; Ming Li; Stefanie Jäger; Jeremy Mason-Herr; et al. Inhibition of a NEDD8 Cascade Restores Restriction of HIV by APOBEC3G. PLOS Pathogens 2012, 8, e1003085, 10.1371/journal.ppat.1003085.
- Tsai-Ling Liao; Chung-Yi Wu; Wen-Chi Su; King-Song Jeng; Michael M C Lai; Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. The EMBO Journal 2010, 29, 3879-3890, 10.1038/emboj.2010.250.
- Ting Pan; Zheng Song; Liyang Wu; Guangyan Liu; Xiancai Ma; Zhilin Peng; Mo Zhou; Liting Liang; Bingfeng Liu; Jun Liu; et al. USP49 potently stabilizes APOBEC3G protein by removing ubiquitin and inhibits HIV-1 replication. eLife 2019, 8, 0, 10.7554/elife.48318.
- Ayaka Okada; Yasumasa Iwatani; APOBEC3G-Mediated G-to-A Hypermutation of the HIV-1 Genome: The Missing Link in Antiviral Molecular Mechanisms. Frontiers in Microbiology 2016, 7, 2027, 10.3389/fmicb.2016.02027.
- Christina M. Ulane; Alex Kentsis; Cristian D. Cruz; Jean-Patrick Parisien; Kristi L. Schneider; Curt M. Horvath; Composition and Assembly of STAT-Targeting Ubiquitin Ligase Complexes: Paramyxovirus V Protein Carboxyl Terminus Is an Oligomerization Domain. Journal of Virology 2005, 79, 10180-10189, 10.1128/jvi.79.16.10180-10189.2005.
- Takayuki Hishiki; Qi’En Han; Kei-Ichiro Arimoto; Kunitada Shimotohno; Tatsuhiko Igarashi; Subhash G. Vasudevan; Youichi Suzuki; Naoki Yamamoto; Interferon-mediated ISG15 conjugation restricts dengue virus 2 replication. Biochemical and Biophysical Research Communications 2014, 448, 95-100, 10.1016/j.bbrc.2014.04.081.
- Chen Zhao; Tien-Ying Hsiang; Rei-Lin Kuo; Robert M. Krug; ISG15 conjugation system targets the viral NS1 protein in influenza A virus–infected cells. Proceedings of the National Academy of Sciences 2010, 107, 2253-2258, 10.1073/pnas.0909144107.
- Florine E.M. Scholte; Marko Zivcec; John V. Dzimianski; Michelle K. Deaton; Jessica R. Spengler; Stephen R. Welch; Stuart T. Nichol; Scott D. Pegan; Christina F. Spiropoulou; Eric Bergeron; et al. Crimean-Congo Hemorrhagic Fever Virus Suppresses Innate Immune Responses via a Ubiquitin and ISG15 Specific Protease. Cell Reports 2017, 20, 2396-2407, 10.1016/j.celrep.2017.08.040.
- Takayuki Abe; Nanae Minami; Rheza Gandi Bawono; Chieko Matsui; Lin Deng; Takasuke Fukuhara; Yoshiharu Matsuura; Ikuo Shoji; ISGylation of Hepatitis C Virus NS5A Protein Promotes Viral RNA Replication via Recruitment of Cyclophilin A. Journal of Virology 2020, 94, 0, 10.1128/jvi.00532-20.
- Nora Schmidt; Patricia Domingues; Filip Golebiowski; Corinna Patzina; Michael H. Tatham; Ronald T. Hay; Benjamin G. Hale; An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proceedings of the National Academy of Sciences 2019, 116, 17399-17408, 10.1073/pnas.1907031116.
- Shu-Chuan Chen; Luan-Yin Chang; Yi-Wei Wang; Yi-Chun Chen; Kuo-Feng Weng; Shin-Ru Shih; Hsiu-Ming Shih; Sumoylation-promoted Enterovirus 71 3C Degradation Correlates with a Reduction in Viral Replication and Cell Apoptosis*. Journal of Biological Chemistry 2011, 286, 31373-31384, 10.1074/jbc.m111.254896.
- Yan Liu; Zhenhua Zheng; Bo Shu; Jin Meng; Yuan Zhang; Caishang Zheng; Xianliang Ke; Peng Gong; Qinxue Hu; Hanzhong Wang; et al. SUMO Modification Stabilizes Enterovirus 71 Polymerase 3D To Facilitate Viral Replication. Journal of Virology 2016, 90, 10472-10485, 10.1128/jvi.01756-16.
- Xiancai Ma; Tao Yang; Yuewen Luo; Liyang Wu; Yawen Jiang; Zheng Song; Ting Pan; Bingfeng Liu; Guangyan Liu; Jun Liu; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, 0, 10.7554/elife.42426.
- Chien-Hung Liu; Ligang Zhou; Guifang Chen; Robert M. Krug; Battle between influenza A virus and a newly identified antiviral activity of the PARP-containing ZAPL protein. Proceedings of the National Academy of Sciences 2015, 112, 14048-14053, 10.1073/pnas.1509745112.
- Chuan Xia; Jennifer J. Wolf; Chuankai Sun; Mengqiong Xu; Caleb J. Studstill; Jun Chen; Hanh Ngo; Hua Zhu; B. Hahm; PARP1 Enhances Influenza A Virus Propagation by Facilitating Degradation of Host Type I Interferon Receptor. Journal of Virology 2020, 94, 0, 10.1128/jvi.01572-19.
- Hao-Sen Chiang; Helene Minyi Liu; The Molecular Basis of Viral Inhibition of IRF- and STAT-Dependent Immune Responses. Frontiers in Immunology 2019, 9, 3086, 10.3389/fimmu.2018.03086.
- Annie Bibeau-Poirier; Simon-Pierre Gravel; Jean-François Clément; Sébastien Rolland; Geneviève Rodier; Philippe Coulombe; John Hiscott; Nathalie Grandvaux; Sylvain Meloche; Marc J. Servant; et al. Involvement of the IκB Kinase (IKK)-Related Kinases Tank-Binding Kinase 1/IKKi and Cullin-Based Ubiquitin Ligases in IFN Regulatory Factor-3 Degradation. The Journal of Immunology 2006, 177, 5059-5067, 10.4049/jimmunol.177.8.5059.
- Henning Hofmann; Thomas D. Norton; Megan L. Schultz; Sylvie B. Polsky; Nicole Sunseri; Nathaniel R. Landau; Inhibition of CUL4A Neddylation Causes a Reversible Block to SAMHD1-Mediated Restriction of HIV-1. Journal of Virology 2013, 87, 11741-11750, 10.1128/jvi.02002-13.
- Haiwei Sun; Wei Yao; Kai Wang; Yingjuan Qian; Antonio Tricoli; Yong-Sam Jung; Inhibition of neddylation pathway represses influenza virus replication and pro-inflammatory responses. Virology 2018, 514, 230-239, 10.1016/j.virol.2017.11.004.
- Robert G. Spiro; Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 2002, 12, 43R-56R, 10.1093/glycob/12.4.43r.
- Ieva Bagdonaite; Hans H Wandall; Global aspects of viral glycosylation. Glycobiology 2018, 28, 443-467, 10.1093/glycob/cwy021.
- David J. Vigerust; Virginia L. Shepherd; Virus glycosylation: role in virulence and immune interactions. Trends in Microbiology 2007, 15, 211-218, 10.1016/j.tim.2007.03.003.
- Yasunori Watanabe; Thomas A. Bowden; Ian A. Wilson; Max Crispin; Exploitation of glycosylation in enveloped virus pathobiology. Biochimica et Biophysica Acta (BBA) - General Subjects 2019, 1863, 1480-1497, 10.1016/j.bbagen.2019.05.012.
- Paula E. Magnelli; Alicia M. Bielik; Ellen P Guthrie; Identification and characterization of protein glycosylation using specific endo- and exoglycosidases. Journal of Visualized Experiments 2011, 10, e3749-e3749, 10.3791/3749.
- Kelley W. Moremen; Michael Tiemeyer; Alison V. Nairn; Vertebrate protein glycosylation: diversity, synthesis and function. Nature Reviews Molecular Cell Biology 2012, 13, 448-462, 10.1038/nrm3383.
- Justine D Mintern; Jose A. Villadangos; Antigen-presenting cells look within during influenza infection. Nature Medicine 2015, 21, 1123-1125, 10.1038/nm.3971.
- Nadine Evan Montfoort; Evelyn Evan Der Aa; Andrea M Woltman; Understanding MHC Class I Presentation of Viral Antigens by Human Dendritic Cells as a Basis for Rational Design of Therapeutic Vaccines. Frontiers in Immunology 2014, 5, 182, 10.3389/fimmu.2014.00182.
- Edward B Irvine; Galit Alter; Understanding the role of antibody glycosylation through the lens of severe viral and bacterial diseases. Glycobiology 2020, 30, 241-253, 10.1093/glycob/cwaa018.
- Giuseppe Lofano; Matthew J. Gorman; Ashraf Siddig Yousif; Wen-Han Yu; Julie M. Fox; Anne-Sophie Dugast; Margaret E. Ackerman; Todd J. Suscovich; Joshua A. Weiner; Dan H. Barouch; et al. Antigen-specific antibody Fc glycosylation enhances humoral immunity via the recruitment of complement. Science Immunology 2018, 3, eaat7796, 10.1126/sciimmunol.aat7796.
- Valerie J. Kinchen; Muhammad N. Zahid; Andrew I. Flyak; Mary G. Soliman; Gerald H. Learn; Shuyi Wang; Edgar Davidson; Benjamin J. Doranz; Stuart C. Ray; Andrea L. Cox; et al. Broadly Neutralizing Antibody Mediated Clearance of Human Hepatitis C Virus Infection. Cell Host & Microbe 2018, 24, 717-730.e5, 10.1016/j.chom.2018.10.012.
- Matthew Zirui Tay; Kevin Wiehe; Justin Pollara; Antibody-Dependent Cellular Phagocytosis in Antiviral Immune Responses. Frontiers in Immunology 2019, 10, 332, 10.3389/fimmu.2019.00332.
- J. S. M. Peiris; J. S. Porterfield; Antibody-mediated enhancement of Flavivirus replication in macrophage-like cell lines. Nature 1979, 282, 509-511, 10.1038/282509a0.
- Wanwisa Dejnirattisai; Piyada Supasa; Wiyada Wongwiwat; Alexander Rouvinski; Giovanna Barba-Spaeth; Thaneeya Duangchinda; Anavaj Sakuntabhai; Van-Mai Cao-Lormeau; Thaneeya Duangchinda Prida Malasit; Alexander Rouvinski Giovanna Barba-Spaeth Felix A Rey; et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nature Immunology 2016, 17, 1102-1108, 10.1038/ni.3515.
- W. K. Eddie Ip; Kwok Hung Chan; Helen K. W. Law; Gloria H. W. Tso; Eric K. P. Kong; Wilfred H. S. Wong; Yuk Fai To; Raymond W. H. Yung; Eudora Y. Chow; Ka Leung Au; et al. Mannose-Binding Lectin in Severe Acute Respiratory Syndrome Coronavirus Infection. Journal of Infectious Diseases 2005, 191, 1697-1704, 10.1086/429631.
- Panisadee Avirutnan; Richard E. Hauhart; Mary A. Marovich; Peter Garred; John P. Atkinson; Michael S. Diamond; Complement-Mediated Neutralization of Dengue Virus Requires Mannose-Binding Lectin. mBio 2011, 2, 0, 10.1128/mbio.00276-11.
- William B. Klimstra; Elizabeth M. Nangle; M. Shane Smith; Andrew D. Yurochko; Kate D. Ryman; DC-SIGN and L-SIGN Can Act as Attachment Receptors for Alphaviruses and Distinguish between Mosquito Cell- and Mammalian Cell-Derived Viruses. Journal of Virology 2003, 77, 12022-12032, 10.1128/jvi.77.22.12022-12032.2003.
- Elena Pokidysheva; Ying Zhang; Anthony J. Battisti; Carol M. Bator-Kelly; Paul R. Chipman; Chuan Xiao; G. Glenn Gregorio; Wayne A. Hendrickson; Richard J Kuhn; Michael G. Rossmann; et al. Cryo-EM Reconstruction of Dengue Virus in Complex with the Carbohydrate Recognition Domain of DC-SIGN. Cell 2006, 124, 485-493, 10.1016/j.cell.2005.11.042.
- Balaji Manicassamy; Jizhen Wang; Haiqing Jiang; Lijun Rong; Comprehensive Analysis of Ebola Virus GP1 in Viral Entry. Journal of Virology 2005, 79, 4793-4805, 10.1128/jvi.79.8.4793-4805.2005.
- Olivier Reynard; Malgorzata Borowiak; Valentina A. Volchkova; Sebastien Delpeut; Mathieu Mateo; Viktor E. Volchkov; Ebolavirus Glycoprotein GP Masks both Its Own Epitopes and the Presence of Cellular Surface Proteins. Journal of Virology 2009, 83, 9596-9601, 10.1128/jvi.00784-09.
- Jason S. McLellan; William C. Ray; Mark E. Peeples; Structure and Function of Respiratory Syncytial Virus Surface Glycoproteins. Current Topics in Microbiology and Immunology 2013, 372, 83-104, 10.1007/978-3-642-38919-1_4.
- J. Li; R. Bhuvanakantham; J. Howe; M.-L. Ng; The glycosylation site in the envelope protein of West Nile virus (Sarafend) plays an important role in replication and maturation processes. Journal of General Virology 2006, 87, 613-622, 10.1099/vir.0.81320-0.
- Changqing Yu; Sunan Li; Xianfeng Zhang; İlyas Khan; Iqbal Ahmad; Yulong Zhou; Shuo Li; Jing Shi; Yu Wang; Yong-Hui Zheng; et al. MARCH8 Inhibits Ebola Virus Glycoprotein, Human Immunodeficiency Virus Type 1 Envelope Glycoprotein, and Avian Influenza Virus H5N1 Hemagglutinin Maturation. mBio 2020, 11, 0, 10.1128/mbio.01882-20.
- Derek L. Carbaugh; Ralph Baric; Helen M. LaZear; Envelope Protein Glycosylation Mediates Zika Virus Pathogenesis. Journal of Virology 2019, 93, 0, 10.1128/jvi.00113-19.
- Dongming Zhao; Libin Liang; Shuai Wang; Tomomi Nakao; Yanbing Li; Liling Liu; Yuntao Guan; Satoshi Fukuyama; Zhigao Bu; Yoshihiro Kawaoka; et al. Glycosylation of the Hemagglutinin Protein of H5N1 Influenza Virus Increases Its Virulence in Mice by Exacerbating the Host Immune Response. Journal of Virology 2017, 91, 0, 10.1128/jvi.02215-16.
- Michelle A. Nelson; Lara J. Herrero; Jason A. L. Jeffery; Marion Hoehn; Penny A. Rudd; Aroon Supramaniam; Brian H. Kay; Peter A. Ryan; Suresh Mahalingam; Role of envelope N-linked glycosylation in Ross River virus virulence and transmission. Journal of General Virology 2016, 97, 1094-1106, 10.1099/jgv.0.000412.
- Marie Flamand; Françoise Megret; Magali Mathieu; Jean Lepault; Félix A. Rey; Vincent Deubel; Dengue Virus Type 1 Nonstructural Glycoprotein NS1 Is Secreted from Mammalian Cells as a Soluble Hexamer in a Glycosylation-Dependent Fashion. Journal of Virology 1999, 73, 6104-6110, 10.1128/jvi.73.7.6104-6110.1999.
- Somchai Thiemmeca; Chamaiporn Tamdet; Nuntaya Punyadee; Tanapan Prommool; Adisak Songjaeng; Sansanee Noisakran; Chunya Puttikhunt; John P. Atkinson; Michael S. Diamond; Alongkot Ponlawat; et al. Secreted NS1 Protects Dengue Virus from Mannose-Binding Lectin–Mediated Neutralization. The Journal of Immunology 2016, 197, 4053-4065, 10.4049/jimmunol.1600323.
- Karen P. Coss; Snezana Vasiljevic; Laura K. Pritchard; Stefanie A. Krumm; Molly Glaze; Sharon Madzorera; Penny L. Moore; Max Crispin; Katie Doores; HIV-1 Glycan Density Drives the Persistence of the Mannose Patch within an Infected Individual. Journal of Virology 2016, 90, 11132-11144, 10.1128/jvi.01542-16.
- P.J. Klasse; Gabriel Ozorowski; Rogier W. Sanders; P J Klasse; Env Exceptionalism: Why Are HIV-1 Env Glycoproteins Atypical Immunogens?. Cell Host & Microbe 2020, 27, 507-518, 10.1016/j.chom.2020.03.018.
- Kshitij Wagh; Edward F. Kreider; Yingying Li; Hannah J. Barbian; Gerald H. Learn; Elena Giorgi; Peter T. Hraber; Timothy G. Decker; Andrew G. Smith; Marcos V. Gondim; et al. Completeness of HIV-1 Envelope Glycan Shield at Transmission Determines Neutralization Breadth. Cell Reports 2018, 25, 893-908.e7, 10.1016/j.celrep.2018.09.087.
- Michael S. Cohen; Paul Chang; Insights into the biogenesis, function, and regulation of ADP-ribosylation. Nature Chemical Biology 2018, 14, 236-243, 10.1038/nchembio.2568.
- Michael O. Hottiger; Paul O Hassa; Bernhard Lüscher; Herwig Schüler; Friedrich Koch-Nolte; Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends in Biochemical Sciences 2010, 35, 208-219, 10.1016/j.tibs.2009.12.003.
- Matthew E. Grunewald; Anthony R. Fehr; Jeremiah Athmer; Stanley Perlman; The coronavirus nucleocapsid protein is ADP-ribosylated. Virology 2018, 517, 62-68, 10.1016/j.virol.2017.11.020.
- Péter Bai; Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Molecular Cell 2015, 58, 947-958, 10.1016/j.molcel.2015.01.034.
- Anthony R. Fehr; Sasha A. Singh; Catherine M. Kerr; Shin Mukai; Hideyuki Higashi; Masanori Aikawa; The impact of PARPs and ADP-ribosylation on inflammation and host–pathogen interactions. Genes & Development 2020, 34, 341-359, 10.1101/gad.334425.119.
- Svetlana Atasheva; Maryna Akhrymuk; Elena I. Frolova; Ilya Frolov; New PARP Gene with an Anti-Alphavirus Function. Journal of Virology 2012, 86, 8147-8160, 10.1128/jvi.00733-12.
- Denisse A. Gutiérrez; Luis Valdes; Ché Serguera; Manuel Llano; Poly(ADP-ribose) polymerase-1 silences retroviruses independently of viral DNA integration or heterochromatin formation.. Journal of General Virology 2016, 97, 1686-1692, 10.1099/jgv.0.000466.
- Murilo T. D. Bueno; Daniel Reyes; Luis Valdes; Adarsh Saheba; Eduardo Urias; Crystal Mendoza; Oliver I. Fregoso; Manuel Llano; Poly(ADP-Ribose) Polymerase 1 Promotes Transcriptional Repression of Integrated Retroviruses. Journal of Virology 2012, 87, 2496-2507, 10.1128/jvi.01668-12.
- Lili Li; Hui Zhao; Ping Liu; Chunfeng Li; Natalie Quanquin; Xue Ji; Nina Sun; Peishuang Du; Cheng-Feng Qin; Ning Lu; et al. PARP12suppresses Zika virus infection through PARP-dependent degradation of NS1 and NS3 viral proteins. Science Signaling 2018, 11, eaas9332, 10.1126/scisignal.aas9332.
- Yiping Zhu; Guifang Chen; Fengxiang Lv; Xinlu Wang; Xin Ji; Yihui Xu; Jing Sun; L. Wu; Yong-Tang Zheng; Guangxia Gao; et al. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proceedings of the National Academy of Sciences 2011, 108, 15834-15839, 10.1073/pnas.1101676108.
- Julie A. Kerns; Michael Emerman; Harmit S. Malik; Positive Selection and Increased Antiviral Activity Associated with the PARP-Containing Isoform of Human Zinc-Finger Antiviral Protein. PLOS Genetics 2008, 4, e21, 10.1371/journal.pgen.0040021.
- Johannes Schwerk; Frank W. Soveg; Andrew P. Ryan; Kerri R. Thomas; Lauren D. Hatfield; Snehal Ozarkar; Adriana Forero; Alison M. Kell; Justin A. Roby; Lomon So; et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nature Immunology 2019, 20, 1610-1620, 10.1038/s41590-019-0527-6.
- Tingting Guo; Yibo Zuo; Liping Qian; Jin Liu; Yukang Yuan; Kailin Xu; Ying Miao; Qian Feng; Xiangjie Chen; Lincong Jin; et al. ADP-ribosyltransferase PARP11 modulates the interferon antiviral response by mono-ADP-ribosylating the ubiquitin E3 ligase β-TrCP. Nature Microbiology 2019, 4, 1872-1884, 10.1038/s41564-019-0428-3.
- Hélène Malet; Bruno Coutard; Saïd Jamal; Hélène Dutartre; Nicolas Papageorgiou; Maarit Neuvonen; Tero Ahola; Naomi Forrester; Ernest A. Gould; Daniel Lafitte; et al. The Crystal Structures of Chikungunya and Venezuelan Equine Encephalitis Virus nsP3 Macro Domains Define a Conserved Adenosine Binding Pocket. Journal of Virology 2009, 83, 6534-6545, 10.1128/jvi.00189-09.
- Mohammad K. Parvez; The hepatitis E virus ORF1 ‘X-domain’ residues form a putative macrodomain protein/Appr-1″-pase catalytic-site, critical for viral RNA replication. Gene 2015, 566, 47-53, 10.1016/j.gene.2015.04.026.
- Matthew E. Grunewald; Yating Chen; Chad V. Kuny; Takashi Maejima; Robert Lease; Dana Ferraris; Masanori Aikawa; Christopher S. Sullivan; Stanley Perlman; Anthony R. Fehr; et al. The coronavirus macrodomain is required to prevent PARP-mediated inhibition of virus replication and enhancement of IFN expression. PLOS Pathogens 2019, 15, e1007756, 10.1371/journal.ppat.1007756.
- Hyo Chol Ha; Krishna Juluri; Yan Zhou; Steve Leung; Monika Hermankova; Solomon H. Snyder; Poly(ADP-ribose) polymerase-1 is required for efficient HIV-1 integration. Proceedings of the National Academy of Sciences 2001, 98, 3364-3368, 10.1073/pnas.051633498.
- Changqing Li; Yannick Debing; Gytis Jankevicius; Johan Neyts; Ivan Ahel; Bruno Coutard; Bruno Canard; Viral Macro Domains Reverse Protein ADP-Ribosylation. Journal of Virology 2016, 90, 8478-8486, 10.1128/jvi.00705-16.
- Rachy Abraham; Debra Hauer; Robert Lyle McPherson; Age Utt; Ilsa T. Kirby; Michael S. Cohen; Andres Merits; Anthony K L Leung; Diane E. Griffin; ADP-ribosyl–binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proceedings of the National Academy of Sciences 2018, 115, E10457-E10466, 10.1073/pnas.1812130115.
- Anthony R. Fehr; Gytis Jankevicius; Ivan Ahel; Stanley Perlman; Viral Macrodomains: Unique Mediators of Viral Replication and Pathogenesis. Trends in Microbiology 2018, 26, 598-610, 10.1016/j.tim.2017.11.011.
- Hong Jiang; Xiaoyu Zhang; Xiao Chen; Pornpun Aramsangtienchai; Zhen Tong; Hening Lin; Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chemical Reviews 2017, 118, 919-988, 10.1021/acs.chemrev.6b00750.
- Sharon L. Swierczynski; Perry J. Blackshear; Myristoylation-dependent and Electrostatic Interactions Exert Independent Effects on the Membrane Association of the Myristoylated Alanine-rich Protein Kinase C Substrate Protein in Intact Cells. Journal of Biological Chemistry 1996, 271, 23424-23430, 10.1074/jbc.271.38.23424.
- Y. Kuroda; N. Suzuki; T. Kataoka; The effect of posttranslational modifications on the interaction of Ras2 with adenylyl cyclase. Science 1993, 259, 683-686, 10.1126/science.8430318.
- Justyna Sobocińska; Paula Roszczenko-Jasińska; Anna Ciesielska; Katarzyna Kwiatkowska; Protein Palmitoylation and Its Role in Bacterial and Viral Infections. Frontiers in Immunology 2018, 8, 0, 10.3389/fimmu.2017.02003.
- Cheryl A. Keller; Xu Yuan; Patrizia Panzanelli; Michelle L. Martin; Melissa J Alldred; Marco Sassoè-Pognetto; Bernhard Lüscher; The gamma 2 Subunit of GABAA Receptors Is a Substrate for Palmitoylation by GODZ. The Journal of Neuroscience 2004, 24, 5881-5891, 10.1523/jneurosci.1037-04.2004.
- M. F. Schmidt; M. Bracha; M. J. Schlesinger; Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins.. Proceedings of the National Academy of Sciences 1979, 76, 1687-1691, 10.1073/pnas.76.4.1687.
- Michael F.G. Schmidt; Milton J. Schlesinger; Fatty acid binding to vesicular stomatitis virus glycoprotein: a new type of post-translational modification of the viral glycoprotein. Cell 1979, 17, 813-819, 10.1016/0092-8674(79)90321-0.
- Julie Claudinon; Pauline Gonnord; Emilie Beslard; Marta Marchetti; Keith Mitchell; Cédric Boularan; Ludger Johannes; Pierre Eid; Christophe Lamaze; Palmitoylation of Interferon-α (IFN-α) Receptor Subunit IFNAR1 Is Required for the Activation of Stat1 and Stat2 by IFN-α. Journal of Biological Chemistry 2009, 284, 24328-24340, 10.1074/jbc.m109.021915.
- Charles C. Bailey; Guocai Zhong; I-Chueh Huang; Michael Farzan; IFITM-Family Proteins: The Cell's First Line of Antiviral Defense. Annual Review of Virology 2014, 1, 261-283, 10.1146/annurev-virology-031413-085537.
- Jocelyn C. Hach; Temet McMichael; Nicholas M. Chesarino; Jacob S. Yount; Palmitoylation on Conserved and Nonconserved Cysteines of Murine IFITM1 Regulates Its Stability and Anti-Influenza A Virus Activity. Journal of Virology 2013, 87, 9923-9927, 10.1128/jvi.00621-13.
- Jacob S Yount; Bruno Moltedo; Yu-Ying Yang; Guillaume Charron; Thomas M Moran; Carolina B López; Howard C. Hang; Palmitoylome profiling reveals S-palmitoylation–dependent antiviral activity of IFITM3. Nature Chemical Biology 2010, 6, 610-614, 10.1038/nchembio.405.
- Michael L. Grantham; Wai-Hong Wu; Erin N. Lalime; Maria E. Lorenzo; Sabra L. Klein; Andrew Pekosz; Palmitoylation of the Influenza A Virus M2 Protein Is Not Required for Virus Replication In Vitro but Contributes to Virus Virulence. Journal of Virology 2009, 83, 8655-8661, 10.1128/jvi.01129-09.
- Itay Rousso; Mark B. Mixon; Benjamin K. Chen; Peter S. Kim; Palmitoylation of the HIV-1 envelope glycoprotein is critical for viral infectivity. Proceedings of the National Academy of Sciences 2000, 97, 13523-13525, 10.1073/pnas.240459697.
- Jolene Ramsey; Emily C. Renzi; Randy J. Arnold; Jonathan C. Trinidad; Suchetana Mukhopadhyay; Palmitoylation of Sindbis Virus TF Protein Regulates Its Plasma Membrane Localization and Subsequent Incorporation into Virions. Journal of Virology 2016, 91, 0, 10.1128/jvi.02000-16.
- Corrin E. McBride; Carolyn E. Machamer; Palmitoylation of SARS-CoV S protein is necessary for partitioning into detergent-resistant membranes and cell–cell fusion but not interaction with M protein. Virology 2010, 405, 139-148, 10.1016/j.virol.2010.05.031.
- Michael Veit; Palmitoylation of virus proteins. Biology of the Cell 2012, 104, 493-515, 10.1111/boc.201200006.
- Ming-Jhan Wu; Saravanabalaji Shanmugam; Christoph Welsch; MinKyung Yi; Palmitoylation of Hepatitis C Virus NS2 Regulates Its Subcellular Localization and NS2-NS3 Autocleavage. Journal of Virology 2019, 94, 0, 10.1128/jvi.00906-19.
- Jérôme Gouttenoire; Angela Pollán; Laurence Abrami; Noémie Oechslin; Johann Mauron; Maxime Matter; Joël Oppliger; Dagmara Szkolnicka; Viet Loan Dao Thi; F. Gisou Van Der Goot; et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLOS Pathogens 2018, 14, e1007471, 10.1371/journal.ppat.1007471.
- William Bakhache; Aymeric Neyret; Eric Bernard; Andres Merits; Laurence Briant; Palmitoylated Cysteines in Chikungunya Virus nsP1 Are Critical for Targeting to Cholesterol-Rich Plasma Membrane Microdomains with Functional Consequences for Viral Genome Replication. Journal of Virology 2020, 94, 0, 10.1128/jvi.02183-19.
- Sebastian Maurer-Stroh; Frank Eisenhaber; Myristoylation of viral and bacterial proteins. Trends in Microbiology 2004, 12, 178-185, 10.1016/j.tim.2004.02.006.
- Thomas Strecker; Anna Maisa; Stephane Daffis; Robert Eichler; Oliver Lenz; Wolfgang Garten; The role of myristoylation in the membrane association of the Lassa virus matrix protein Z. Virology Journal 2006, 3, 93-93, 10.1186/1743-422X-3-93.
- San Suwanmanee; Yuvadee Mahakhunkijcharoen; Sumate Ampawong; Pornsawan Leaungwutiwong; Dorothée Missé; Natthanej Luplertlop; Inhibition ofN‐myristoyltransferase1 affects dengue virus replication. MicrobiologyOpen 2019, 8, e00831, 10.1002/mbo3.831.
- San Suwanmanee; Yuvadee Mahakhunkijcharoen; Sumate Ampawong; Pornsawan Leaungwutiwong; Dorothée Missé; Natthanej Luplertlop; Inhibition of N‐myristoyltransferase1 affects dengue virus replication. MicrobiologyOpen 2019, 8, e00831, 10.1002/mbo3.831.
- Irena Corbic Ramljak; Julia Stanger; Antonio Real-Hohn; Dominik Dreier; Laurin Wimmer; Monika Redlberger-Fritz; Wolfgang Fischl; Karin Klingel; Marko D. Mihovilovic; Dieter Blaas; et al. Cellular N-myristoyltransferases play a crucial picornavirus genus-specific role in viral assembly, virion maturation, and infectivity. PLOS Pathogens 2018, 14, e1007203, 10.1371/journal.ppat.1007203.
- Lauren O'neil; Kathryn Andenoro; Isabella Pagano; Laura Carroll; Leah Langer; Zachary Dell; Davina Perera; Bradley W. Treece; Frank Heinrich; Mathias Lösche; et al. HIV-1 matrix-31 membrane binding peptide interacts differently with membranes containing PS vs. PI(4,5)P2. Biochimica et Biophysica Acta (BBA) - Biomembranes 2016, 1858, 3071-3081, 10.1016/j.bbamem.2016.09.010.
- Jiaming Cao; Meng Qu; Hongtao Liu; Xuan Wan; Fang Li; Ali Hou; Yan Zhou; Bo Sun; Linjun Cai; Weiheng Su; et al. Myristoylation of EV71 VP4 is Essential for Infectivity and Interaction with Membrane Structure. Virologica Sinica 2020, 35, 599-613, 10.1007/s12250-020-00226-1.
- H G Kräusslich; C Hölscher; Q Reuer; J Harber; E Wimmer; Myristoylation of the poliovirus polyprotein is required for proteolytic processing of the capsid and for viral infectivity.. Journal of Virology 1990, 64, 2433-2436, 10.1128/jvi.64.5.2433-2436.1990.
- Fang L. Zhang; Patrick J. Casey; Protein Prenylation: Molecular Mechanisms and Functional Consequences. Annual Review of Biochemistry 1996, 65, 241-269, 10.1146/annurev.bi.65.070196.001325.
- Matthew J. Bick; John-William N. Carroll; Guangxia Gao; Stephen P. Goff; Charles M. Rice; Margaret R. Macdonald; Expression of the Zinc-Finger Antiviral Protein Inhibits Alphavirus Replication. Journal of Virology 2003, 77, 11555-11562, 10.1128/jvi.77.21.11555-11562.2003.
- Chunfu Wang; Michael Gale; Brian C. Keller; Hua Huang; Michael S. Brown; Joseph L. Goldstein; Jin Ye; Identification of FBL2 As a Geranylgeranylated Cellular Protein Required for Hepatitis C Virus RNA Replication. Molecular Cell 2005, 18, 425-434, 10.1016/j.molcel.2005.04.004.
- James C. Otto; Patrick J. Casey; The Hepatitis Delta Virus Large Antigen Is Farnesylated Both in Vitro and in Animal Cells. Journal of Biological Chemistry 1996, 271, 4569-4572, 10.1074/jbc.271.9.4569.
- C Z Lee; P J Chen; Ding-Shinn Chen; Large hepatitis delta antigen in packaging and replication inhibition: role of the carboxyl-terminal 19 amino acids and amino-terminal sequences.. Journal of Virology 1995, 69, 5332-5336, 10.1128/jvi.69.9.5332-5336.1995.
- Shigao Yang; Alfred T. Harding; Catherine Sweeney; David Miao; Gregory Swan; Connie Zhou; Zhaozhao Jiang; Katherine A. Fitzgerald; Gianna Hammer; Martin O. Bergo; et al. Control of antiviral innate immune response by protein geranylgeranylation. Science Advances 2019, 5, eaav7999, 10.1126/sciadv.aav7999.
- Fuxiao Xin; Predrag Radivojac; Post-translational modifications induce significant yet not extreme changes to protein structure. Bioinformatics 2012, 28, 2905-2913, 10.1093/bioinformatics/bts541.
- Fatima Ardito; Michele Giuliani; Donatella Perrone; Giuseppe Troiano; L. Lo Muzio; The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). International Journal of Molecular Medicine 2017, 40, 271-280, 10.3892/ijmm.2017.3036.
- Juan Liu; Cheng Qian; Xuetao Cao; Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15-30, 10.1016/j.immuni.2016.06.020.
- Weinan Zheng; Shuaishuai Cao; Can Chen; Jing Li; Shuang Zhang; Jingwen Jiang; Yange Niu; Wenhui Fan; Yun Li; Yuhai Bi; et al. Threonine 80 phosphorylation of non-structural protein 1 regulates the replication of influenza A virus by reducing the binding affinity with RIG-I. Cellular Microbiology 2016, 19, e12643, 10.1111/cmi.12643.
- Oscar A. Tarazona; Olivier Pourquié; Exploring the Influence of Cell Metabolism on Cell Fate through Protein Post-translational Modifications. Developmental Cell 2020, 54, 282-292, 10.1016/j.devcel.2020.06.035.
- C S Rubin; O M Rosen; Protein Phosphorylation. Annual Review of Biochemistry 1975, 44, 831-887, 10.1146/annurev.bi.44.070175.004151.
- Forrest Keck; Pouya Ataey; Moushimi Amaya; Charles L. Bailey; Aarthi Narayanan; Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies. Viruses 2015, 7, 5257-5273, 10.3390/v7102872.
- Kate M. Franz; William J. Neidermyer; Yee-Joo Tan; Sean P. J. Whelan; Jonathan C. Kagan; STING-dependent translation inhibition restricts RNA virus replication. Proceedings of the National Academy of Sciences 2018, 115, E2058-E2067, 10.1073/pnas.1716937115.
- Yang Li; Heather L. Wilson; Endre Kiss-Toth; Regulating STING in health and disease. Journal of Inflammation 2017, 14, 1-21, 10.1186/s12950-017-0159-2.
- Zhe Ma; Blossom Damania; The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host & Microbe 2016, 19, 150-158, 10.1016/j.chom.2016.01.010.
- Omer Abid Kathum; Tobias Schräder; Darisuren Anhlan; Carolin Nordhoff; Swantje Liedmann; Amit Pande; Alexander Mellmann; Christina Ehrhardt; Viktor Wixler; Stephan Ludwig; et al. Phosphorylation of influenza A virus NS1 protein at threonine 49 suppresses its interferon antagonistic activity. Cellular Microbiology 2016, 18, 784-791, 10.1111/cmi.12559.
- Lok Man J. Law; Jason C. Everitt; Martin D. Beatch; Charles F. B. Holmes; Tom C. Hobman; Phosphorylation of Rubella Virus Capsid Regulates Its RNA Binding Activity and Virus Replication. Journal of Virology 2003, 77, 1764-71, 10.1128/jvi.77.3.1764-1771.2003.
- Helena Vihinen; Juhani Saarinen; Phosphorylation Site Analysis of Semliki Forest Virus Nonstructural Protein 3. Journal of Biological Chemistry 2000, 275, 27775-27783, 10.1074/jbc.m002195200.
- Bastian Thaa; Roberta Biasiotto; Kai Eng; Maarit Neuvonen; Benjamin Götte; Lara Rheinemann; Margit Mutso; Age Utt; Finny Varghese; Giuseppe Balistreri; et al. Differential Phosphatidylinositol-3-Kinase-Akt-mTOR Activation by Semliki Forest and Chikungunya Viruses Is Dependent on nsP3 and Connected to Replication Complex Internalization. Journal of Virology 2015, 89, 11420-11437, 10.1128/jvi.01579-15.
- Lin Zhu; Ting Gao; Weihong Yang; Yaoning Liu; Xuan Liu; Yong Hu; Yanwen Jin; Ping Li; Ke Xu; Gang Zou; et al. Ebola virus replication is regulated by the phosphorylation of viral protein VP35.. Biochemical and Biophysical Research Communications 2020, 521, 687-692, 10.1016/j.bbrc.2019.10.147.
- Christopher M. Ziegler; Philip Eisenhauer; Inessa Manuelyan; Marion E. Weir; Emily A. Bruce; Bryan A. Ballif; Jason W. Botten; Host-Driven Phosphorylation Appears to Regulate the Budding Activity of the Lassa Virus Matrix Protein. Pathogens 2018, 7, 97, 10.3390/pathogens7040097.
- Michela Mazzon; Meleri Jones; Andrew Davidson; Benjamin Chain; Michael Jacobs; Dengue Virus NS5 Inhibits Interferon‐α Signaling by Blocking Signal Transducer and Activator of Transcription 2 Phosphorylation. The Journal of Infectious Diseases 2009, 200, 1261-1270, 10.1086/605847.
- Juliet Morrison; Maudry Laurent-Rolle; Ana M. Maestre; Ricardo Rajsbaum; Giuseppe Pisanelli; Viviana Simon; Lubbertus C. F. Mulder; Ana Fernandez-Sesma; Adolfo García-Sastre; Dengue Virus Co-opts UBR4 to Degrade STAT2 and Antagonize Type I Interferon Signaling. PLOS Pathogens 2013, 9, e1003265, 10.1371/journal.ppat.1003265.
- Stephanie Thurmond; Boxiao Wang; Jikui Song; Rong Hai; Suppression of Type I Interferon Signaling by Flavivirus NS5. Viruses 2018, 10, 712, 10.3390/v10120712.
- Yingchun Zeng; Linbai Ye; Shengli Zhu; Hong Zheng; Peng Zhao; Weijia Cai; Liya Su; Yinglong She; Zhenghui Wu; The nucleocapsid protein of SARS-associated coronavirus inhibits B23 phosphorylation. Biochemical and Biophysical Research Communications 2008, 369, 287-291, 10.1016/j.bbrc.2008.01.096.
- Jingfang Mu; Yaohui Fang; Qi Yang; Ting Shu; An Wang; Muhan Huang; Liang Jin; Fei Deng; Yang Qiu; Xi Zhou; et al. SARS-CoV-2 N protein antagonizes type I interferon signaling by suppressing phosphorylation and nuclear translocation of STAT1 and STAT2. Cell Discovery 2020, 6, 1-4, 10.1038/s41421-020-00208-3.
- Milan Surjit; Ravinder Kumar; Rabi N. Mishra; Malireddy K. Reddy; Vincent T. K. Chow; Sunil K. Lal; The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Protein Is Phosphorylated and Localizes in the Cytoplasm by 14-3-3-Mediated Translocation. Journal of Virology 2005, 79, 11476-11486, 10.1128/jvi.79.17.11476-11486.2005.
- Edward M. Kennedy; David G. Courtney; Kevin Tsai; Bryan R. Cullen; Viral Epitranscriptomics. Journal of Virology 2017, 91, e02263-16, 10.1128/jvi.02263-16.
- Laura Marcos-Villar; Juan Díaz-Colunga; Juan Sandoval; Noelia Zamarreño; Sara Landeras-Bueno; Manel Esteller; Ana Falcón; Amelia Nieto; Epigenetic control of influenza virus: role of H3K79 methylation in interferon-induced antiviral response. Scientific Reports 2018, 8, 1-13, 10.1038/s41598-018-19370-6.
- Zheng Zhang; Bryan C. Nikolai; Leah A. Gates; Sung Yun Jung; Edward B. Siwak; Zhang Zheng; Andrew P. Rice; Bert W. O’Malley; Qin Feng; Crosstalk between histone modifications indicates that inhibition of arginine methyltransferase CARM1 activity reverses HIV latency. Nucleic Acids Research 2017, 45, 9348-9360, 10.1093/nar/gkx550.
- Ying Liu; Yinghua Niu; Lu Li; Khalid A. Timani; Victor L. He; Chris Sanburns; Jiafeng Xie; Johnny J. He; Tat expression led to increased histone 3 tri-methylation at lysine 27 and contributed to HIV latency in astrocytes through regulation of MeCP2 and Ezh2 expression. Journal of NeuroVirology 2019, 25, 508-519, 10.1007/s13365-019-00751-0.
- Rachel Van Duyne; Rebecca Easley; Weilin Wu; Reem Berro; Caitlin Pedati; Zachary Klase; Kylene Kehn-Hall; Elizabeth K Flynn; David E. Symer; Fatah Kashanchi; et al. Lysine methylation of HIV-1 Tat regulates transcriptional activity of the viral LTR. Retrovirology 2008, 5, 40-13, 10.1186/1742-4690-5-40.
- Naoki Sakane; Hye-Sook Kwon; Sara Pagans; Katrin Kaehlcke; Yasuhiro Mizusawa; Masafumi Kamada; Kara G. Lassen; Jonathan Chan; Warner C. Greene; Martina Schnoelzer; et al. Activation of HIV Transcription by the Viral Tat Protein Requires a Demethylation Step Mediated by Lysine-specific Demethylase 1 (LSD1/KDM1). PLOS Pathogens 2011, 7, e1002184, 10.1371/journal.ppat.1002184.
- Jiaoyu Shan; Binbin Zhao; Zhao Shan; Jia Nie; Rong Deng; Rui Xiong; Andy Tsun; Weiqi Pan; Hanzhi Zhao; Ling Chen; et al. Histone demethylase LSD1 restricts influenza A virus infection by erasing IFITM3-K88 monomethylation. PLOS Pathogens 2017, 13, e1006773, 10.1371/journal.ppat.1006773.
- Laura A. Murray; Ashton N. Combs; Pranav Rekapalli; Ileana M. Cristea; Methods for characterizing protein acetylation during viral infection.. Methods in Enzymology 2019, 626, 587-620, 10.1016/bs.mie.2019.06.030.
- Matloob Husain; Chen-Yi Cheung; Histone Deacetylase 6 Inhibits Influenza A Virus Release by Downregulating the Trafficking of Viral Components to the Plasma Membrane via Its Substrate, Acetylated Microtubules. Journal of Virology 2014, 88, 11229-11239, 10.1128/jvi.00727-14.
- Dai Hatakeyama; Masaki Shoji; Seiya Yamayoshi; Rina Yoh; Naho Ohmi; Shiori Takenaka; Ayaka Saitoh; Yumie Arakaki; Aki Masuda; Tsugunori Komatsu; et al. Influenza A virus nucleoprotein is acetylated by histone acetyltransferases PCAF and GCN5. Journal of Biological Chemistry 2018, 293, 7126-7138, 10.1074/jbc.ra117.001683.
- Cesar Muñoz-Fontela; Dolores González; Laura Marcos-Villar; Michela Campagna; Pedro Gallego; José González-Santamaría; Daniel Herranz; Wei Gu; Manuel Serrano; Stuart A. Aaronson; et al. Acetylation is indispensable for p53 antiviral activity. Cell Cycle 2011, 10, 3701-3705, 10.4161/cc.10.21.17899.
- Samuel A Williams; Lin-Feng Chen; Hakju Kwon; Carmen M Ruiz-Jarabo; Eric Verdin; Warner C. Greene; NF-κB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. The EMBO Journal 2005, 25, 139-149, 10.1038/sj.emboj.7600900.
- Su Jin Choi; Hyun‐Cheol Lee; Jae‐Hoon Kim; Song Yi Park; Tae‐Hwan Kim; Woon‐Kyu Lee; Duk‐Jae Jang; Ji‐Eun Yoon; Young‐Il Choi; Seihwan Kim; et al. HDAC 6 regulates cellular viral RNA sensing by deacetylation of RIG ‐I. The EMBO Journal 2016, 35, 429-442, 10.15252/embj.201592586.
- Huan Chen; Yingjuan Qian; Xin Chen; Zhiyang Ruan; Yuetian Ye; Hongjun Chen; Lorne A. Babiuk; Yong-Sam Jung; Jianjun Dai; HDAC6 Restricts Influenza A Virus by Deacetylation of the RNA Polymerase PA Subunit. Journal of Virology 2018, 93, 0, 10.1128/jvi.01896-18.
- Jingjiao Ma; Rujuan Wu; GuanLong Xu; Yuqiang Cheng; Zhaofei Wang; Hengan Wang; Yaxian Yan; Jinxiang Li; Jianhe Sun; Acetylation at K108 of the NS1 protein is important for the replication and virulence of influenza virus.. Veterinary Research 2020, 51, 20-10, 10.1186/s13567-020-00747-3.
- Sebastian Giese; Kevin Ciminski; Hardin Bolte; Étori Aguiar Moreira; Seema Lakdawala; Zehan Hu; Quinnlan David; Larissa Kolesnikova; Veronika Götz; Yongxu Zhao; et al. Role of influenza A virus NP acetylation on viral growth and replication. Nature Communications 2017, 8, 1-11, 10.1038/s41467-017-01112-3.
- Jie Zan; Song Liu; Dong-Nan Sun; Kai-Kun Mo; Yan Yan; Juan Liu; Bo-Li Hu; Jin-Yan Gu; Min Liao; Ji-Yong Zhou; et al. Rabies Virus Infection Induces Microtubule Depolymerization to Facilitate Viral RNA Synthesis by Upregulating HDAC6. Frontiers in Cellular and Infection Microbiology 2017, 7, 146, 10.3389/fcimb.2017.00146.
- Qi Yang; Jielin Tang; Rongjuan Pei; Xiaoxiao Gao; Jing Guo; Chonghui Xu; Yun Wang; Qian Wang; Chunchen Wu; Yuan Zhou; et al. Host HDAC4 regulates the antiviral response by inhibiting the phosphorylation of IRF3. Journal of Molecular Cell Biology 2018, 11, 158-169, 10.1093/jmcb/mjy035.
- Catherine Eichwald; Francesca Arnoldi; Andrea S. Laimbacher; Elisabeth M. Schraner; Cornel Fraefel; Peter Wild; Oscar R. Burrone; Mathias Ackermann; Rotavirus Viroplasm Fusion and Perinuclear Localization Are Dynamic Processes Requiring Stabilized Microtubules. PLoS ONE 2012, 7, e47947, 10.1371/journal.pone.0047947.
- John S. L. Parker; Teresa J. Broering; Jonghwa Kim; Darren E. Higgins; Max L. Nibert; Reovirus Core Protein μ2 Determines the Filamentous Morphology of Viral Inclusion Bodies by Interacting with and Stabilizing Microtubules. Journal of Virology 2002, 76, 4483-4496, 10.1128/jvi.76.9.4483-4496.2002.
- Qiuqin Feng; Zhonglan Su; Shiyu Song; Hui Xu; Bin Zhang; Long Yi; Man Tian; Hongwei Wang; Histone deacetylase inhibitors suppress RSV infection and alleviate virus-induced airway inflammation. International Journal of Molecular Medicine 2016, 38, 812-822, 10.3892/ijmm.2016.2691.
- Chien-Yi Lu; Yi-Chih Chang; Chun-Hung Hua; Chieh Chuang; Su-Hua Huang; Szu-Hao Kung; Mann-Jen Hour; Cheng-Wen Lin; Tubacin, an HDAC6 Selective Inhibitor, Reduces the Replication of the Japanese Encephalitis Virus via the Decrease of Viral RNA Synthesis. International Journal of Molecular Sciences 2017, 18, 954, 10.3390/ijms18050954.
- H G Kräusslich; C Hölscher; Q Reuer; J Harber; E Wimmer; Myristoylation of the poliovirus polyprotein is required for proteolytic processing of the capsid and for viral infectivity.. Journal of Virology 1990, 64, 2433-2436, 10.1128/jvi.64.5.2433-2436.1990.