Protein aggregation is classically considered the main cause of neuronal death in neurodegenerative diseases (NDDs). However, increasing evidence suggests that alteration of RNA metabolism is a key factor in the etiopathogenesis of these complex disorders. Non-coding RNAs are the major contributor to the human transcriptome and are particularly abundant in the central nervous system, where they have been proposed to be involved in the onset and development of NDDs. Interestingly, some ncRNAs (such as lncRNAs, circRNAs and pseudogenes) share a common functionality in their ability to regulate gene expression by modulating miRNAs in a phenomenon known as the competing endogenous RNA mechanism. Moreover, ncRNAs are found in body fluids where their presence and concentration could serve as potential non-invasive biomarkers of NDDs.
- competing endogenous RNAs (ceRNA)
- neurodegenerative diseases (NDDs)
- extracellular/circulating biomarkers
- microRNA
- long non-coding RNA
- circular RNA
- pseudogene
- mRNA
- ceRNA network (ceRNET)
- RNA editing
1. Introduction
ncRNAs can be classified into two groups according to their length: small ncRNAs (<200 nucleotides) and long ncRNAs (>200 nucleotides) [1]. Among small ncRNAs, microRNAs (miRNA) stand out, being around 22 nucleotides long and regulating gene expression at the post-transcriptional level in a sequence-specific manner [2]. Approximately 70% of the identified miRNAs are expressed in the brain [3] and have been described as major regulators of neuronal homeostasis, their misregulation being associated with pathological conditions of CNS [2]. The largest class of ncRNAs in the mammalian genome is long ncRNAs (lncRNAs), which can be further grouped into linear RNAs and circular RNAs [1] [4]. Linear lncRNAs (hereon referred to as lncRNAs) are similar to protein-coding messenger RNA (mRNA) in sequence length and transcriptional and post-transcriptional behavior [1]. However, lncRNAs play a different cellular role compared to mRNAs. Moreover, they have been described to be involved in brain development, neuronal function, maintenance and differentiation [5]. Circular RNAs (circRNAs) represent a relatively recently discovered class of RNAs that, unlike linear RNAs, are characterized by a covalent bond that joins the 5′ and 3′ ends and confers increased stability (half-life of 48 h vs. 10 h for mRNAs) [6]. circRNAs are highly abundant in the brain, enriched in synaptoneurosomes and upregulated during neuronal differentiation [7], so they could be promising biomarkers in age-associated NDDs.
On the other hand, a considerable number of pseudogenes can be transcribed to ncRNAs, even though they have historically been regarded as inactive gene sequences [8] [9]. In fact, there is mounting evidence that pseudogenes may modulate the expression of parental as well as unrelated genes [8] [9]. Therefore, alteration of pseudogene transcription could perturb gene expression homeostasis leading to disease [8].
In 2011, Pier Paolo Pandolfi’s group proposed the so-called ceRNA hypothesis [10], which sought to explain how RNAs “talk” to each other, establishing interactions that modify functional genetic information and that may play major roles in pathological conditions. This hypothesis is based on the fact that miRNAs can recognize their specific target sites called miRNA response elements (MRE) in different RNA molecules, causing target repression via miRNA-RISC complex-mediated degradation. Thereby, miRNAs could mediate regulatory crosstalk between the diverse components of the transcriptome, comprising mRNAs and ncRNAs, which include pseudogenes, lncRNAs and circRNAs.
In a simplified manner, when two RNA molecules share the same MRE they potentially compete for the same pool of miRNAs. Thus, when the expression of a ceRNA is upregulated, it will bind and titrate more miRNAs (phenomenon called miRNA sponging), leaving fewer miRNA molecules available for binding the mRNA with shared MRE. Hence, this corresponding mRNA will become derepressed. In reverse, when the ceRNA levels are reduced as a consequence of a biological disturbance, the corresponding mRNA will be downregulated due to hyperrepression (Figure 1).
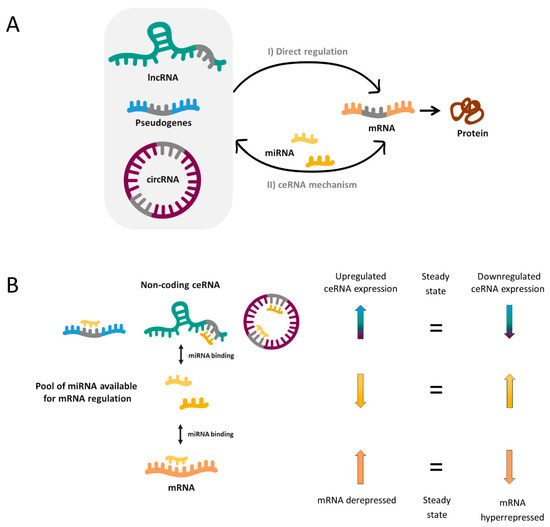
Without doubt, the reality is more complex and a miRNA can bind more than one mRNA (50% of miRNAs are predicted to target 1–400 mRNAs and some of them up to 1000) [11]. Likewise, most ceRNAs contain 1 to 10 MREs [11] and, as a consequence, complex ceRNA networks involving a large number of RNA molecules are established. Novel bioinformatic and computational tools have enabled to elucidate an increasing number of ceRNA networks, as well as predict the most important enclaves of them. These may provide a valuable global vision to identify new biomarkers, underlying pathways or potential therapeutic targets for complex disorders such as NDDs.
2. ceRNA Networks and Neurodegenerative Diseases
Over the last years, the ceRNA hypothesis has been corroborated by a large number of experiments. However, investigation of ceRNA mechanisms and their interaction networks has been mainly carried out in cancer research [12] [13] [14] [15]. Nevertheless, some advances have also been made in the field of NDDs (Table 1).
Table 1. miRNA-ceRNAs networks experimentally validated associated with NDDs
|
Disease |
ncRNA |
miRNA |
mRNA |
Sample |
Ref. |
|
|
AD
|
lncRNA |
BACE1-AS |
miR-29, miR-485, miR-761,miR-124 and miR-107 |
BACE1 |
Computational analysis from human data and cellular and mouse models |
[16] |
|
|
miR-214-3p |
- |
[17] |
|||
|
|
miR-132-3p |
- |
[18] |
|||
|
XIST |
miR-124 |
BACE1 |
Cellular and mouse models |
[19] |
||
|
|
miR-132 |
- |
[20] |
|||
|
NEAT1 |
miR-124 |
BACE1 |
Cellular and mouse models |
[21] |
||
|
|
miR-107 |
- |
[22] |
|||
|
SOX21-AS1 |
miR-107 |
- |
Cellular model |
[23] |
||
|
NEAT1 HOTAIR MALAT1 |
miR-107, miR-103, miR-16, miR-195, miR-15a and miR-15b |
CDK5R1 |
Cellular model
|
[24] |
||
|
MALAT1 |
miR-125b |
CDK5, FOXQ1 and PTGS2 |
Cellular and rat models |
[25] |
||
|
|
miR-30b |
CNR1 |
[26] |
|||
|
TUG1 |
miR-15a |
ROCK1 |
Cellular and mouse models |
[27] |
||
|
SNHG1 |
miR-137 |
KREMEN1 |
Cellular model and human primary cell culture |
[28] |
||
|
|
miR-361-3p |
ZNF217 |
[29] |
|||
|
lncRNA-ATB |
miR-200 |
ZNF217 |
Cellular model |
[30] |
||
|
LINC00094 |
miR‐224‐4p miR‐497‐5p |
SH3GL2 |
Cellular model |
[31] |
||
|
MIAT |
miR-150-5p |
VEGF |
Cellular and mouse models |
[32] |
||
|
Rpph1 |
miR-326 |
PKM2 |
Cellular and mouse models |
[33] |
||
|
|
miR-122 |
Wnt1 |
[34] |
|||
|
|
miR-330-5p |
CDC42 |
[35] |
|||
|
linc00507 |
miR-181c-5p |
MAPT TTBK1 |
Cellular and mouse models |
[36] |
||
|
lnc-ANRIL |
mir-125a |
TNF-α, IL1B IL6 and IL17 |
Cellular model |
[37] |
||
|
circRNA |
ciRS-7 |
miR-7 |
UBE2A |
Human brain |
[38] |
|
|
|
*miR-7 |
*NF-Κb/p65 |
Cellular models |
|||
|
circ_0000950 |
miR-103 |
PTGS2 |
Cellular models |
[42] |
||
|
circHDAC9 |
miR-138 |
Sirt1 |
Cellular and mouse models |
[43] |
||
|
|
miR-142-5p |
- |
[44] |
|||
|
PD |
pseudogene |
GBAP1 |
miR-22-3p |
GBA |
Cellular models |
[45] |
|
lncRNA |
SNHG1 |
miR-153-3p miR-15b-5p miR-7 miR-221/222 |
PTEN SIAH1, GSK3β NLRP3 CDKN1B (p27) |
Cellular and mouse models |
[46] [49] [50] |
|
|
HAGLROs |
miR-100 |
ATG10 |
Cellular and mouse models |
[51] |
||
|
HOTAIR |
miR-874-5p |
ATG10 |
Cellular and mouse models |
[52] |
||
|
|
miR-126-5p |
RAB3IP |
[53] |
|||
|
NEAT1 |
miR-212-5p |
RAB3IP |
Cellular models |
[54] |
||
|
|
miR-1277-5p |
ARHGAP26 |
[55] |
|||
|
|
miR-124 |
- |
[56] |
|||
|
AL049437 |
miR-205-5p |
MAPK1 |
Cellular and mouse models |
[57] |
||
|
MALAT1 |
miR-205-5p |
LRRK2 |
Cellular and mouse models
|
[58] |
||
|
|
miR-124 |
DAPK1 |
||||
|
|
miR-129 |
SNCA (α-syn) |
[61] |
|||
|
SNHG14 |
miR-133b |
SNCA |
Cellular and mouse models |
[62] |
||
|
LincRNA-p21 |
miR-1277-5p |
SNCA |
Cellular and mouse models |
[63] |
||
|
|
miR-181 family |
PRKCD (PKC-δ) |
[64] |
|||
|
|
miR-625 |
TRPM2 |
[65] |
|||
|
GAS5 |
miR-223-3p |
NLRP3 |
Cellular and mouse models |
[66] |
||
|
BDNF-AS |
miR-125b-5p |
- |
Cellular and mouse models |
[67] |
||
|
Mirt2 |
miR-101 |
- |
Cellular model |
[68] |
||
|
lncRNA H19 |
miR-301b-3p |
HPRT1 |
Computational analysis from human data and cellular and mouse models |
[69] |
||
|
|
miR-585-3p |
PIK3R3 |
[70] |
|||
|
circRNA |
*ciRS-7 |
miR-7 |
SNCA |
Cellular and mouse models |
||
|
circSNCA |
miR-7 |
SNCA |
Cellular model |
[76] |
||
|
circzip-2 |
*miR-60 |
M60.4ZK470.2, igeg-2 and idhg-1 |
Worm model |
[77] |
||
|
circDLGAP4 |
miR-134-5p |
CREB |
Cellular and mouse models |
[78] |
||
|
MS |
lncRNA |
Gm15575 |
miR-686 |
CCL7 |
Cellular and mouse models |
[79] |
|
PVT1 |
miR-21-5p |
SOCS5 |
Cellular and mouse models |
[80] |
||
|
TUG |
miR-9-5p |
NFKB1 (p50) |
Cellular and mouse models |
[81] |
||
|
HOTAIR |
miR-136-5p |
AKT2 |
Cellular and mouse models |
[82] |
||
|
GAS5 |
miR-137 |
- |
Human blood |
[83] |
||
|
circRNA |
hsa_circ_0106803 |
*miR-149 |
*ASIC1a |
Human blood (PMBCs) |
||
|
hsa_circ_0005402 hsa_circ_0035560 |
*14 miRNAs (miR-1248, miR-766) |
- |
Human blood (PMBCs) |
[86] |
||
|
SCA7 |
lncRNA |
lnc-SCA7 |
miR-124 |
ATXN7 |
Human samples, and cellular and animal models |
[87] |
* Experimental validation is needed.
3. RNA Editing Alteration and ceRNA Networks
RNA editing is an important mechanism of post-transcriptional processing that can modify RNA molecules by altering its sequences through insertion, deletion, or conversion of a nucleotide [88] [89]. Recent discoveries suggest that RNA editing critically regulates neurodevelopment and normal neuronal function, for which some crucial aspects of neurodegenerative diseases may stem from the modification of both coding and non-coding RNA [89] [90] [91].
The most common type of RNA editing is the conversion of adenosine to inosine (A-to-I), in which enzymes encoded by the adenosine deaminase acting on RNA (ADAR) gene family catalyze the deamination of adenosine (A) nucleotides to inosines (I) [92]. Critical consequences are derived from this modification, since inosine (I) is interpreted by the translation and splicing machineries as guanosine (G) [93]. Editing of pre-mRNA coding regions can lead to codon change that may result in increased diversity of protein isoforms and their respective function [88]. However, most of the RNA editing happens in non-coding RNAs, which can affect their stability, biogenesis and target recognition [88] [93] [94]. In fact, it has been reported that ADAR is involved in circRNA biogenesis by editing and destabilizing the flanking Alu repeat sequences, which makes circRNA production less favorable [94] [95]. Moreover, editing events can affect both the maturation and the expression of miRNAs, but if the modification occurs in MREs or in miRNA seed regions (regions in miRNA sequence that largely determine the binding specificity on its targets), the spectrum of miRNA targets, or “targetome”, shall be changed [96]. Therefore, a single editing site in an RNA molecule could drastically modify its function, resulting in new or different ceRNA networks that regulate gene expression.
Interestingly, A-to-I editing has been reported specifically reduced in SALS motor neurons due to the progressive downregulation of ADAR2 [97] [98]. Based on this evidence, Hosaka et al. [99] searched for extracellular RNAs with ADAR2-dependent A-to-I sites that may reflect the intracellular pathological process and thus could be potentially good ALS biomarkers. A total of six RNAs were identified. Among these, a circRNA (hsa_circ_0125620, also called circGRIA2) with an ADAR2-dependent site was detected in human SH-SY5Y neuroblastoma cells as well as in their culture medium [99]. Therefore, variations in RNA editing efficiency in ALS, as a consequence of decreased ADAR2 activity, could be potentially measured in peripheral circRNAs and other relatively stable ncRNAs. In light of this evidence, this editing phenomenon may be considered a very important aspect, since it allows obtain relevant information of disease pathological process from non-coding RNAs.
Other NDDs, such as AD and PD, also present alterations in RNA editing patterns [100] [90] [101] [102]. In fact, a recent study has explored how RNA editing in AD contributes to the regulation of AD-related processes in blood cells in two populations of patients [103]. Results identified differentially edited sites predicted to disrupt miRNA target sites in five genes. In all cases, decreased editing was observed in AD suggesting a greater miRNA-binding affinity relative to controls [103]. In light of this evidence, alterations in RNA editing could result in a specific RNA profile, given by different amount of RNAs, modified interaction networks and editing levels or efficiencies changes in A-to-I sites, that could be useful to identify new robust biomarkers of these NDDs (Figure 2).
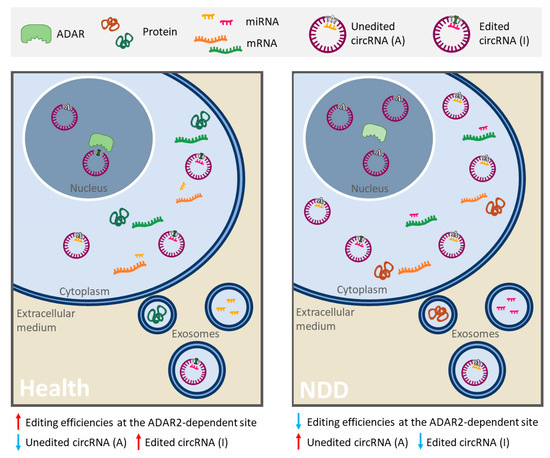
Figure 2. Schematic representation of alterations in RNA editing that could provide a specific RNA profile in neurodegenerative diseases (NDDs). In some linear and circular RNAs, the enzyme ADAR2 deaminates adenosine (A) into inosine (I), resulting in important biological consequences (especially in ncRNAs). On the one hand, a single editing site in MRE or miRNA seed region can drastically change its set of targets. In this image, circ-Purple acts as a miR-Yellow sponge, which regulates the mRNA expression of Orange gene (right panel). Deamination of A into I in circ-Purple could affect its binding site for miR-Yellow. In consequence, circ-Purple stops sponging miR-Yellow, and it may bind to another miRNA (miR-Pink) and promote the expression of Green gene (left panel). Hence, the ceRNA interaction network has changed, emerging a new or different regulatory axis. On the other hand, ADAR editing negatively regulates circRNA biogenesis, resulting in a decrease of circRNA levels (in the left panel there is less circ-Purple expression than in the right panel). In NDDs with a diminution of A-to-I RNA editing (like ALS, AD or PD), a different and opposite profile/pattern could be observed (right panel) with respect to a normal editing efficiency of ADAR (left panel). Therefore, alterations mediated by RNA editing in RNAs and its ceRNA interaction networks may serve as robust biomarkers of these NDDs. This figure is based on a previously published figure [99] [104].
4. Conclusion
The vast majority of NDDs can be definitively diagnosed only after death or in advanced stage, and their previous diagnosis is based on ruling out other possible causes for the symptoms. For most NDDs, there is no cure or treatment capable of reversing the damage due to neuronal death. Therefore, it is critical to find new biomarkers that would facilitate an early diagnosis, prognosis and efficient monitoring of therapeutic interventions.
In the search for new biomarkers, non-coding RNAs have been proposed as promising tools for diagnosis and prognosis. Many ncRNAs often arise from genes that cause NDDs or are somehow involved in the development of one of these disorders (like BACE1-AS or circSNCA). Thus, ceRNETs established by these ncRNAs could well be, at least in some cases, disease and even stage-specific. However, as reported in this review, ncRNAs are commonly misregulated in several NDDs (Figure 3). This is the case, for example, of the lncRNAs SNHG1 and HOTAIR, which are altered in AD [28] [29] and PD [46] [47] [48] [49] [50], and PD [52] [53] and MS [82], respectively. However, their miRNA targets may vary depending on cell types affected by the disease and, therefore, the mechanism of action may also differ. Similarly, miR-7 has been shown sponged by ciRS-7/CDR1as and circSNCA in AD [38] [39] [40] [41] and PD [76], respectively, being detrimental in the first case and beneficial in the second, due to regulation of different target mRNAs. The apparent discrepancy between the anti and pro cell death activity of miR-7 reflects the complex regulatory role of miRNAs, so further research is required to clarify their function in different cellular and disease contexts.
In this way, by analyzing various elements of the altered ceRNETs, it may be possible to differentiate one NDD from another even if there were common components. Ideally, working with several correlatable molecular targets at the same time (lncRNAs/circRNAs/pseudogenes-miRNA-mRNAs) increases the sensitivity and reliability of ceRNETs as biomarkers. It should be noted that ceRNETs construction also contributes to the identification of new molecular mechanisms of gene regulation that may lead to a better understanding of the etiopathogenesis of the diverse NDDs, as well as to reveal new therapeutic targets and obtain relevant information about the pathological processes of the disease.
In this sense, ceRNETs may also reflect the editing efficiencies of ADAR, a post-transcriptional phenomenon dysregulated in several NDDs. RNA editing can affect the levels and the efficiency of RNA interaction networks, so its alterations could provide a specific RNA fingerprint that helps in the diagnosis or prognosis of NDDs. Finally, the described crosstalk between the RNA molecules in certain ceRNETs is relatively conserved between species, paving the way for translation of data obtained from animal models into clinical practice [105] [106].
Among the main advantages of ceRNETs for biomarker research, the fact that these ncRNAs are easily accessible is noteworthy, since they are extremely stable in circulation and may be detected in exosomes. Such is the case for circRNA CDR1as/ciRS-7 and lncRNA MALAT1, found in exosomes. Interestingly, levels of ciRS-7 in these vesicles depend on the intracellular abundance of the miRNA that it sponges (miR-7) [107]. Furthermore, ciRS-7 and MALAT1 may regulate miRNA expression in target cells after exosomal delivery modulating their phenotype, since these ceRNAs retain their biological activity [107] [108]. Therefore, ciRS-7 and MALAT1 together with other circulating ncRNAs (e.g., NEAT1, GAS5, hsa_circ_061346, hsa_circ_000843) represent promising candidates for peripheral ceRNA biomarkers of NDDs. Although many of the ncRNAs discussed earlier have not been reported in exosomes to date, some of them are predicted to be detected in human blood exosomes by exoRBase (e.g., circSLC8A1, circCORO1C, SNHG1, BACE1-AS) [109]. Indeed, it has recently been demonstrated that plasma exosomal BACE1‑AS levels could serve as a biomarker of AD [110] [111].
Because ceRNA interaction networks are multifactorial, they may represent an advantage in studies of these complex neurodegenerative disorders, one being at the level of biomarkers (combined RNA biomarkers panels) and another at the level of therapeutic targets (modulate the levels of multiple disease-associated RNAs at once by just targeting one).
Nevertheless, it must be taken into account that there is still much to do, since these networks are very complex and their interactions must be experimentally defined [105]. In this sense, some “non-canonical” aspects of ncRNAs have also been described: i) circRNAs that can also sponge or serve as a decoy for RBPs or lncRNAs, ii) miRNAs that may increase the expression of target genes, iii) lncRNAs that can be precursors of smaller ncRNAs and can regulate miRNA and circRNA biogenesis, iv) miRNAs that can direct Ago2 to degrade lncRNA and circRNA, v) lncRNAs that compete with miRNAs for the target site of mRNA, and vi) context-specific miRNA function and target identification [112] [113] [114] [115] [116] [117] [118] [119].
Although the full extent of ceRNA networks still needs to be still determined, the competition of ncRNA and mRNAs for miRNAs constitutes a key point of gene regulation that could underlie some pathological aspects of neurodegenerative diseases, favoring at the end the identification of specific pathological mechanisms for each disease.
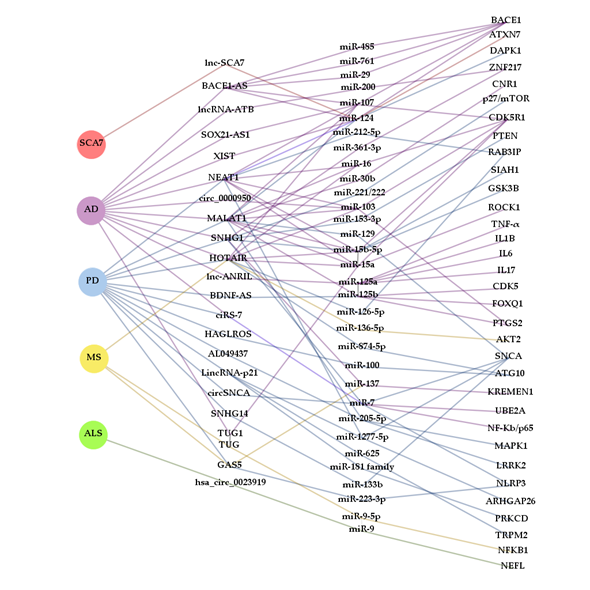
Figure 3. Complexity and interaction of ceRNETs in NDDs. The diagram was constructed with Gephi software from ceRNAs (lncRNAs and circRNAs) that, according to the bibliography cited in this review, contribute to the pathogenesis of more than one neurodegenerative disease and miRNAs that are part of ceRNETs from more than one ceRNA. Interactions between RNA molecules are represented with lines colored in accordance with the NDD background they have been described in: spinocerebellar ataxia type 7 (SCA7) (red), Alzheimer’s disease (AD) (purple), Parkinson’s disease (PD) (blue), multiple sclerosis (MS) (yellow) and amyotrophic lateral sclerosis (ALS) (green).
This entry is adapted from the peer-reviewed paper 10.3390/ijms21249582
References
- Noorul Amin; Annette McGrath; Yi-Ping Phoebe Chen; Evaluation of deep learning in non-coding RNA classification. Nature Machine Intelligence 2019, 1, 246-256, 10.1038/s42256-019-0051-2.
- Sean Quinlan; Aidan Kenny; Miguel Medina; Tobias Engel; Eva M Jimenez-Mateos; MicroRNAs in Neurodegenerative Diseases. Int. Rev. Cell Mol. Biol. 2017, 334, 309-343, .
- Xinwei Cao; Gene Yeo; Alysson R. Muotri; Tomoko Kuwabara; Fred H. Gage; NONCODING RNAS IN THE MAMMALIAN CENTRAL NERVOUS SYSTEM. Annual Review of Neuroscience 2006, 29, 77-103, 10.1146/annurev.neuro.29.051605.112839.
- Jia Jia Chan; Yvonne Tay; Noncoding RNA: RNA Regulatory Networks in Cancer.. Int. J. Mol. Sci. 2018, 19, 1310, .
- Chang-Wei Wei; Ting Luo; Shan-Shan Zou; An-Shi Wu; The Role of Long Noncoding RNAs in Central Nervous System and Neurodegenerative Diseases. Frontiers in Behavioral Neuroscience 2018, 12, 175, 10.3389/fnbeh.2018.00175.
- William R. Jeck; Jessica A. Sorrentino; Kai Wang; Michael K. Slevin; Christin E. Burd; Jinze Liu; William F. Marzluff; Norman E. Sharpless; Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141-157, 10.1261/rna.035667.112.
- Agnieszka Rybak-Wolf; Christin Stottmeister; Petar Glažar; Marvin Jens; Natalia Pino; Sebastian Giusti; Mor Hanan; Mikaela Behm; Osnat Bartok; Reut Ashwal-Fluss; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Molecular Cell 2015, 58, 870-885, 10.1016/j.molcel.2015.03.027.
- Xingyi Guo; Mingyan Lin; Shira Rockowitz; Herbert M. Lachman; Deyou Zheng; Characterization of Human Pseudogene-Derived Non-Coding RNAs for Functional Potential. PLOS ONE 2014, 9, e93972, 10.1371/journal.pone.0093972.
- Baikang Pei; Cristina Sisu; Adam Frankish; Cédric Howald; Lukas Habegger; Xinmeng Jasmine Mu; Rachel Harte; Suganthi Balasubramanian; Andrea Tanzer; Mark Diekhans; et al. The GENCODE pseudogene resource. Genome Biology 2012, 13, R51-26, 10.1186/gb-2012-13-9-r51.
- Leonardo Salmena; Laura Poliseno; Yvonne Tay; Lev Kats; Pier Paolo Pandolfi; A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language?. Cell 2011, 146, 353-358, 10.1016/j.cell.2011.07.014.
- Ugo Ala; Florian A. Karreth; Carla Bosia; Andrea Pagnani; Riccardo Taulli; Valentine Léopold; Yvonne Tay; Paolo Provero; Riccardo Zecchina; Pier Paolo Pandolfi; et al. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proceedings of the National Academy of Sciences 2013, 110, 7154-7159, 10.1073/pnas.1222509110.
- Rasoul Abdollahzadeh; Abdolreza Daraei; Yaser Mansoori; Masoumeh Sepahvand; Mahsa M. Amoli; Javad Tavakkoly-Bazzaz; Competing endogenous RNA (ceRNA) cross talk and language in ceRNA regulatory networks: A new look at hallmarks of breast cancer. Journal of Cellular Physiology 2018, 234, 10080-10100, 10.1002/jcp.27941.
- Yanyan Liu; Jingyu Zhu; Xiaoli Ma; Shuyi Han; Dongjie Xiao; Yanfei Jia; Yunshan Wang; ceRNA network construction and comparison of gastric cancer with or without Helicobacter pylori infection. Journal of Cellular Physiology 2018, 234, 7128-7140, 10.1002/jcp.27467.
- Juan Xu; Yongsheng Li; Jianping Lu; Tao Pan; Na Ding; Zishan Wang; Tingting Shao; Jinwen Zhang; Lihua Wang; Xia Li; et al. The mRNA related ceRNA–ceRNA landscape and significance across 20 major cancer types. Nucleic Acids Research 2015, 43, 8169-8182, 10.1093/nar/gkv853.
- Xiaolong Qi; Da-Hong Zhang; Nan Wu; Jun-Hua Xiao; Xiang Wang; Wang Ma; ceRNA in cancer: possible functions and clinical implications. Journal of Medical Genetics 2015, 52, 710-718, 10.1136/jmedgenet-2015-103334.
- Tao Zeng; Haitao Ni; Yue Yu; Mingke Zhang; Minjuan Wu; Qiaoling Wang; Liujun Wang; Sha Xu; Zhenyu Xu; Chen Xu; et al. BACE1-AS prevents BACE1 mRNA degradation through the sequestration of BACE1-targeting miRNAs. Journal of Chemical Neuroanatomy 2019, 98, 87-96, 10.1016/j.jchemneu.2019.04.001.
- Wei He; Songyuan Chi; Xing Jin; Jieyu Lu; Wei Zheng; Jie Yan; Duo Zhang; Long Non-Coding RNA BACE1-AS Modulates Isoflurane-Induced Neurotoxicity to Alzheimer’s Disease Through Sponging miR-214-3p. Neurochemical Research 2020, 45, 2324-2335, 10.1007/s11064-020-03091-2.
- Yunli Ge; Xiaolin Song; Jianfeng Liu; Chun Liu; Changshui Xu; The Combined Therapy of Berberine Treatment with lncRNA BACE1-AS Depletion Attenuates Aβ25–35 Induced Neuronal Injury Through Regulating the Expression of miR-132-3p in Neuronal Cells. Neurochemical Research 2020, 45, 741-751, 10.1007/s11064-019-02947-6.
- Du Yue; Gao Guanqun; Li Jingxin; Suo Sen; Liu Shuang; Sun Yan; Zhang Minxue; Yin Ping; Lu Chong; Zhang Zhuobo; et al. Silencing of long noncoding RNA XIST attenuated Alzheimer's disease‐related BACE1 alteration through miR‐124. Cell Biology International 2019, 44, 630-636, 10.1002/cbin.11263.
- Xueyin Wang; Chao Wang; Caihong Geng; Kunpeng Zhao; LncRNA XIST knockdown attenuates Aβ25-35-induced toxicity, oxidative stress, and apoptosis in primary cultured rat hippocampal neurons by targeting miR-132.. Int. J. Clin. Exp. Pathol. 2018, 11, 3915-3924, .
- Mei-Ying Zhao; Gui-Qing Wang; Ni-Ni Wang; Qiao-Yan Yu; Rong-Li Liu; Wen-Qian Shi; The long-non-coding RNA NEAT1 is a novel target for Alzheimer’s disease progression via miR-124/BACE1 axis. Neurological Research 2019, 41, 489-497, 10.1080/01616412.2018.1548747.
- Sha Ke; Zhaohui Yang; Fei Yang; XiaoMing Wang; Juan Tan; Bo Liao; Long Noncoding RNA NEAT1 Aggravates Aβ-Induced Neuronal Damage by Targeting miR-107 in Alzheimer's Disease. Yonsei Medical Journal 2019, 60, 640-650, 10.3349/ymj.2019.60.7.640.
- Wanru Xu; Kai Li; Qian Fan; Biyun Zong; Ling Han; Knockdown of long non-coding RNA SOX21-AS1 attenuates amyloid-β-induced neuronal damage by sponging miR-107. Bioscience Reports 2020, 40, 1-12, 10.1042/bsr20194295.
- Marco Spreafico; Barbara Grillo; F. Rusconi; Elena Battaglioli; Marco Venturin; Multiple Layers of CDK5R1 Regulation in Alzheimer’s Disease Implicate Long Non-Coding RNAs. International Journal of Molecular Sciences 2018, 19, 2022, 10.3390/ijms19072022.
- Peizhi Ma; Yuanlong Li; Wei Zhang; Fengqin Fang; Jun Sun; Mingzhou Liu; Kun Li; Lingfang Dong; Long Non-coding RNA MALAT1 Inhibits Neuron Apoptosis and Neuroinflammation While Stimulates Neurite Outgrowth and Its Correlation With MiR-125b Mediates PTGS2, CDK5 and FOXQ1 in Alzheimer's Disease. Current Alzheimer Research 2019, 16, 596-612, 10.2174/1567205016666190725130134.
- Li Li; Yuelong Xu; Meng Zhao; Zhiqiang Gao; Neuro-protective roles of long non-coding RNA MALAT1 in Alzheimer's disease with the involvement of the microRNA-30b/CNR1 network and the following PI3K/AKT activation. Experimental and Molecular Pathology 2020, 117, 104545, 10.1016/j.yexmp.2020.104545.
- Xia Li; Sheng-Wu Wang; Xi-Ling Li; Feng-Yuan Yu; Hai-Ming Cong; Knockdown of long non-coding RNA TUG1 depresses apoptosis of hippocampal neurons in Alzheimer’s disease by elevating microRNA-15a and repressing ROCK1 expression. Inflammation Research 2020, 69, 897-910, 10.1007/s00011-020-01364-8.
- Hui Wang; Bo Lu; Jie Chen; Knockdown of lncRNA SNHG1 attenuated Ab25-35 -inudced neuronal injury via regulating KREMEN1 by acting as a ceRNA of miR-137 in neuronal cells. Biochem. Biophys. Res. Commun. 2019, 518, 438-444, .
- Yiwen Gao; Nan Zhang; Chunmei Lv; Na Li; Xueqin Li; Weiwei Li; LncRNA SNHG1 Knockdown Alleviates Amyloid-B-Induced Neuronal Injury by Regulating ZNF217 via Sponging miR-361-3p in Alzheimer’s Disease. J. Alzheimer Dis. 2020, 77, 85-98, .
- Jindong Wang; Tiantian Zhou; Tian Wang; Bailing Wang; Suppression of lncRNA-ATB prevents amyloid-β-induced neurotoxicity in PC12 cells via regulating miR-200/ZNF217 axis. Biomedicine & Pharmacotherapy 2018, 108, 707-715, 10.1016/j.biopha.2018.08.155.
- Lu Zhu; Meiqing Lin; Jun Ma; Wenjing Liu; Lili Gao; Shanshan Wei; Yixue Xue; Xiuli Shang; The role of LINC 00094/miR‐224‐5p (miR‐497‐5p)/Endophilin‐1 axis in Memantine mediated protective effects on blood‐brain barrier in AD microenvironment. Journal of Cellular and Molecular Medicine 2019, 23, 3280-3292, 10.1111/jcmm.14214.
- Qin Jiang; Kun Shan; Xiao Qun-Wang; Rong-Mei Zhou; Hong Yang; Chang Liu; Yu-Jie Li; Jin Yao; Xiu-Miao Li; Yi Shen; et al. Long non-coding RNA-MIAT promotes neurovascular remodeling in the eye and brain. Oncotarget 2016, 7, 49688–49698, .
- Ran Gu; Rui Liu; Lu Wang; Man Tang; Shi-Rong Li; Xiao Hu; LncRNA RPPH1 attenuates Aβ25-35-induced endoplasmic reticulum stress and apoptosis in SH-SY5Y cells via miR-326/PKM2. International Journal of Neuroscience 2020, , , 10.1080/00207454.2020.1746307.
- Ran Gu; Lu Wang; Man Tang; Shi-Rong Li; Rui Liu; Xiao Hu; LncRNA Rpph1 protects amyloid-β induced neuronal injury in SK-N-SH cells via miR-122/Wnt1 axis. International Journal of Neuroscience 2019, 130, 443-453, 10.1080/00207454.2019.1692834.
- Yifei Cai; Ziling Sun; Huizhen Jia; Hongxue Luo; Xiaoyang Ye; Qi Wu; Yi Xiong; Wei Zhang; Jun Wan; Rpph1 Upregulates CDC42 Expression and Promotes Hippocampal Neuron Dendritic Spine Formation by Competing with miR-330-5p. Frontiers in Molecular Neuroscience 2017, 10, 27, 10.3389/fnmol.2017.00027.
- Yan Yan; Hua Yan; Ying Teng; Qin Wang; Ping Yang; Le Zhang; Han Cheng; Siwen Fu; Long non‐coding RNA 00507/miRNA‐181c‐5p/TTBK1/MAPT axis regulates tau hyperphosphorylation in Alzheimer's disease. The Journal of Gene Medicine 2020, 22, e3268, 10.1002/jgm.3268.
- Bingling Zhou; Lijuan Li; Xin Qiu; Jiashun Wu; Lei Xu; Wei Shao; Long non-coding RNA ANRIL knockdown suppresses apoptosis and pro-inflammatory cytokines while enhancing neurite outgrowth via binding microRNA-125a in a cellular model of Alzheimer’s disease. Mol. Med. Rep. 2020, 22, 1489–1497, .
- Yuhai Zhao; Peter N. Alexandrov; Vivian Jaber; Walter J. Lukiw; Deficiency in the Ubiquitin Conjugating Enzyme UBE2A in Alzheimer’s Disease (AD) is Linked to Deficits in a Natural Circular miRNA-7 Sponge (circRNA; ciRS-7). Genes 2016, 7, 116, 10.3390/genes7120116.
- Zhemin Shi; Ting Chen; Qingbin Yao; Lina Zheng; Zhen Zhang; Jingzhao Wang; Zhimei Hu; Hongmei Cui; Yawei Han; Xiaohui Han; et al. The circular RNA ci RS ‐7 promotes APP and BACE 1 degradation in an NF ‐κB‐dependent manner. The FEBS Journal 2017, 284, 1096-1109, 10.1111/febs.14045.
- Tingbo Ye; Meihua Yang; Daochao Huang; Xin Wang; Bingqian Xue; Na Tian; Xiaohui Xu; Liming Bao; Huajian Hu; Tiewei Lv; et al. MicroRNA-7 as a potential therapeutic target for aberrant NF-κB-driven distant metastasis of gastric cancer. Journal of Experimental & Clinical Cancer Research 2019, 38, 1-18, 10.1186/s13046-019-1074-6.
- Doo Chul Choi; Yoon-Jee Chae; Savan Kabaria; Amrita Datta Chaudhuri; Mohit Raja Jain; Hong Li; M. Maral Mouradian; Eunsung Junn; MicroRNA-7 Protects against 1-Methyl-4-Phenylpyridinium-Induced Cell Death by Targeting RelA. The Journal of Neuroscience 2014, 34, 12725-12737, 10.1523/jneurosci.0985-14.2014.
- Hui Yang; Huan Wang; Hong Shang; Xiufen Chen; Shiqi Yang; Yang Qu; Jing Ding; Xuling Li; Circular RNA circ_0000950 promotes neuron apoptosis, suppresses neurite outgrowth and elevates inflammatory cytokines levels via directly sponging miR-103 in Alzheimer’s disease. Cell Cycle 2019, 18, 2197-2214, 10.1080/15384101.2019.1629773.
- Yanjun Lu; Lu Tan; Xiong Wang; Circular HDAC9/microRNA-138/Sirtuin-1 Pathway Mediates Synaptic and Amyloid Precursor Protein Processing Deficits in Alzheimer’s Disease. Neuroscience Bulletin 2019, 35, 877-888, 10.1007/s12264-019-00361-0.
- Nan Zhang; Yiwen Gao; Shaoli Yu; Xiaohong Sun; Ke Shen; Berberine attenuates Aβ42-induced neuronal damage through regulating circHDAC9/miR-142-5p axis in human neuronal cells. Life Sciences 2020, 252, 117637, 10.1016/j.lfs.2020.117637.
- Letizia Straniero; Valeria Rimoldi; Maura Samarani; Stefano Goldwurm; Alessio Di Fonzo; Rejko Krüger; Michela Deleidi; Massimo Aureli; Giulia Soldà; Stefano Duga; et al. The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Scientific Reports 2017, 7, 12702, 10.1038/s41598-017-12973-5.
- Jun Zhao; Lijiao Geng; Yong Chen; Chunfang Wu; SNHG1 promotes MPP+-induced cytotoxicity by regulating PTEN/AKT/mTOR signaling pathway in SH-SY5Y cells via sponging miR-153-3p. Biological Research 2020, 53, 1-11, 10.1186/s40659-019-0267-y.
- Na Xie; Jinxing Qi; Shuang Li; Jianzhong Deng; Yuan Chen; Yajun Lian; Upregulated lncRNA small nucleolar RNA host gene 1 promotes 1‐methyl‐4‐phenylpyridinium ion‐induced cytotoxicity and reactive oxygen species production through miR‐15b‐5p/GSK3β axis in human dopaminergic SH‐SY5Y cells. Journal of Cellular Biochemistry 2018, 120, 5790-5801, 10.1002/jcb.27865.
- Yuan Chen; Ya-Jun Lian; Yun-Qing Ma; Chuan-Jie Wu; Ya-Ke Zheng; Nan-Chang Xie; LncRNA SNHG1 promotes α-synuclein aggregation and toxicity by targeting miR-15b-5p to activate SIAH1 in human neuroblastoma SH-SY5Y cells. NeuroToxicology 2018, 68, 212-221, 10.1016/j.neuro.2017.12.001.
- Bingqing Cao; Tao Wang; Qiumin Qu; Tao Kang; Qian Yang; Long Noncoding RNA SNHG1 Promotes Neuroinflammation in Parkinson’s Disease via Regulating miR-7/NLRP3 Pathway. Neuroscience 2018, 388, 118-127, 10.1016/j.neuroscience.2018.07.019.
- Chen Qian; Yongyi Ye; Hengxu Mao; Longping Yao; Xiang Sun; Baoyan Wang; Hongbo Zhang; Linghai Xie; Huan Zhang; Yizhou Zhang; et al. Downregulated lncRNA-SNHG1 enhances autophagy and prevents cell death through the miR-221/222 /p27/mTOR pathway in Parkinson's disease. Experimental Cell Research 2019, 384, 111614, 10.1016/j.yexcr.2019.111614.
- Tao Peng; Xiaoyan Liu; Jingtao Wang; Yu Liu; Zhenqiang Fu; Xingrong Ma; Junmin Li; Guifang Sun; Yangfei Ji; Jingjing Lu; et al. Long noncoding RNA HAGLROS regulates apoptosis and autophagy in Parkinson’s disease via regulating miR-100/ATG10 axis and PI3K/Akt/mTOR pathway activation. Artificial Cells, Nanomedicine, and Biotechnology 2019, 47, 2764-2774, 10.1080/21691401.2019.1636805.
- Jingya Zhao; Hongli Li; Na Chang; LncRNA HOTAIR promotes MPP+-induced neuronal injury in Parkinson’s disease by regulating the miR-874-5p/ATG10 axis. null 2020, 19, 1141-1153, .
- Qiuyu Lin; Sen Hou; Yuyin Dai; Nan Jiang; Yingjie Lin; LncRNA HOTAIR targets miR-126-5p to promote the progression of Parkinson’s disease through RAB3IP. Biological Chemistry 2018, 400, 1217-1228, 10.1515/hsz-2018-0431.
- Ruiguang Liu; Fenlin Li; Weijie Zhao; Long noncoding RNA NEAT1 knockdown inhibits MPP+-induced apoptosis, inflammation and cytotoxicity in SK-N-SH cells by regulating miR-212-5p/RAB3IP axis. Neuroscience Letters 2020, 731, 135060, 10.1016/j.neulet.2020.135060.
- Shufang Zhou; Dan Zhang; Junnan Guo; Zhenzhen Chen; Yong Chen; Junshi Zhang; Deficiency of NEAT1 prevented MPP+-induced inflammatory response, oxidative stress and apoptosis in dopaminergic SK-N-SH neuroblastoma cells via miR-1277-5p/ARHGAP26 axis. Brain Research 2021, 1750, 147156, 10.1016/j.brainres.2020.147156.
- San-Ping Xie; Fan Zhou; Juan Li; Shu-Jie Duan; NEAT1 regulates MPP+-induced neuronal injury by targeting miR-124 in neuroblastoma cells. Neurosci. Lett. 2019, 708, 134340, .
- Liang Zhang; Jingzhong Wang; Qin Liu; Zhiqiang Xiao; Quande Dai; Knockdown of long non-coding RNA AL049437 mitigates MPP+ -induced neuronal injury in SH-SY5Y cells via the microRNA-205-5p/MAPK1 axis. NeuroToxicology 2020, 78, 29-35, 10.1016/j.neuro.2020.02.004.
- Qin Chen; Xiaoyan Huang; Renjie Li; lncRNA MALAT1/miR-205-5p axis regulates MPP+-induced cell apoptosis in MN9D cells by directly targeting LRRK2.. American journal of translational research 2018, 10, 563-572, .
- Wei Liu; Qishun Zhang; Jianlei Zhang; Wujun Pan; Jingya Zhao; Yuming Xu; Long non-coding RNA MALAT1 contributes to cell apoptosis by sponging miR-124 in Parkinson disease. Cell & Bioscience 2017, 7, 1-9, 10.1186/s13578-017-0147-5.
- Yi Lu; Zhongying Gong; Xiaojie Jin; Peng Zhao; Yuting Zhang; Zhiyun Wang; LncRNA MALAT1 targeting miR‐124‐3p regulates DAPK1 expression contributes to cell apoptosis in Parkinson's Disease. Journal of Cellular Biochemistry 2020, 121, 4838-4848, 10.1002/jcb.29711.
- Dongjian Xia; Rubo Sui; Zhuang Zhang; Administration of resveratrol improved Parkinson’s disease‐like phenotype by suppressing apoptosis of neurons via modulating the MALAT1/miR‐129/SNCA signaling pathway. Journal of Cellular Biochemistry 2018, 120, 4942-4951, 10.1002/jcb.27769.
- Li-Min Zhang; Meng-Han Wang; He-Cheng Yang; Tian Tian; Gui-Fang Sun; Yang-Fei Ji; Wen-Tao Hu; Xi Liu; Jian-Ping Wang; Hong Lu; et al. Dopaminergic neuron injury in Parkinson’s disease is mitigated by interfering lncRNA SNHG14 expression to regulate the miR-133b/ α-synuclein pathway. Aging 2019, 11, 9264-9279, 10.18632/aging.102330.
- Xiaonan Xu; Chengle Zhuang; Zimu Wu; Hongyan Qiu; Haixia Feng; Jun Wu; LincRNA-p21 Inhibits Cell Viability and Promotes Cell Apoptosis in Parkinson's Disease through Activating α-Synuclein Expression. BioMed Research International 2018, 2018, 1-10, 10.1155/2018/8181374.
- Yongyi Ye; Xiaozheng He; Fengfei Lu; Hengxu Mao; Zhiyuan Zhu; Longping Yao; Wanxian Luo; Xiang Sun; Baoyan Wang; Chen Qian; et al. A lincRNA-p21/miR-181 family feedback loop regulates microglial activation during systemic LPS- and MPTP- induced neuro inflammation. Cell Death Dis. 2018, 9, 803, .
- Xiu-Ming Ding; Lian-Jiang Zhao; Han-Yong Qiao; Shao-Lan Wu; Xi-Hui Wang; Long non-coding RNA-p21 regulates MPP+-induced neuronal injury by targeting miR-625 and derepressing TRPM2 in SH-SY5Y cells. Chemico-Biological Interactions 2019, 307, 73-81, 10.1016/j.cbi.2019.04.017.
- Wei Xu; Ling Zhang; Yu Geng; Ye Liu; Long noncoding RNA GAS5 promotes microglial inflammatory response in Parkinson's disease by regulating NLRP3 pathway through sponging miR-223-3p. International Immunopharmacology 2020, 85, 106614, 10.1016/j.intimp.2020.106614.
- Yan Fan; Xue Zhao; Kai Lu; Guizhi Cheng; LncRNA BDNF-AS promotes autophagy and apoptosis in MPTP-induced Parkinson’s disease via ablating microRNA-125b-5p. Brain Research Bulletin 2020, 157, 119-127, 10.1016/j.brainresbull.2020.02.003.
- Yingying Han; Chunyang Kang; Mingyang Kang; Wei Quan; Heming Gao; Zhuan Zhong; Long non-coding RNA Mirt2 prevents TNF-α-triggered inflammation via the repression of microRNA-101. International Immunopharmacology 2019, 76, 105878, 10.1016/j.intimp.2019.105878.
- Jingjing Jiang; Xuanyu Piao; Siying Hu; Jingbo Gao; Min Bao; LncRNA H19 diminishes dopaminergic neuron loss by mediating microRNA-301b-3p in Parkinson’s disease via the HPRT1-mediated Wnt/β-catenin signaling pathway. Aging 2020, 12, 8820-8836, 10.18632/aging.102877.
- Yong Zhang; Qiming Xia; Jun Lin; LncRNA H19 Attenuates Apoptosis in MPTP-Induced Parkinson’s Disease Through Regulating miR-585-3p/PIK3R3. Neurochemical Research 2020, 45, 1700-1710, 10.1007/s11064-020-03035-w.
- Eunsung Junn; Kang-Woo Lee; Byeong Seon Jeong; Teresa W. Chan; Joo-Young Im; M. Maral Mouradian; Repression of -synuclein expression and toxicity by microRNA-7. Proceedings of the National Academy of Sciences 2009, 106, 13052-13057, 10.1073/pnas.0906277106.
- Kuei-Yang Hsiao; H Sunny Sun; Shaw-Jenq Tsai; Circular RNA – New member of noncoding RNA with novel functions. Experimental Biology and Medicine 2017, 242, 1136-1141, 10.1177/1535370217708978.
- Thomas B. Hansen; Trine I. Jensen; Bettina Hjelm Clausen; Jesper Bertram Bramsen; Bente Finsen; Christian Kroun Damgaard; Jørgen Kjems; Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384-388, 10.1038/nature11993.
- Lalit Kumar; Shamsuzzama; Rizwanul Haque; Tanvi Baghel; Aamir Nazir; Circular RNAs: the Emerging Class of Non-coding RNAs and Their Potential Role in Human Neurodegenerative Diseases. Molecular Neurobiology 2016, 54, 7224-7234, 10.1007/s12035-016-0213-8.
- Yiye Eshao; Yinghui Chen; Roles of Circular RNAs in Neurologic Disease. Frontiers in Molecular Neuroscience 2016, 9, 25, 10.3389/fnmol.2016.00025.
- Qiuling Sang; Xiaoyang Liu; Libo Wang; Ling Qi; Wenping Sun; Weiyao Wang; Yajuan Sun; Haina Zhang; CircSNCA downregulation by pramipexole treatment mediates cell apoptosis and autophagy in Parkinson’s disease by targeting miR-7. Aging 2018, 10, 1281-1293, 10.18632/aging.101466.
- Lalit Kumar; Shamsuzzama; Pooja Jadiya; Rizwanul Haque; Shikha Shukla; Aamir Nazir; Functional Characterization of Novel Circular RNA Molecule, circzip-2 and Its Synthesizing Gene zip-2 in C. elegans Model of Parkinson’s Disease. Molecular Neurobiology 2018, 55, 6914-6926, 10.1007/s12035-018-0903-5.
- Zhong Feng; Li Zhang; Sa Wang; Qing Hong; Circular RNA circDLGAP4 exerts neuroprotective effects via modulating miR-134-5p/CREB pathway in Parkinson’s disease. Biochemical and Biophysical Research Communications 2020, 522, 388-394, 10.1016/j.bbrc.2019.11.102.
- Zhengying Bian; Wen Lei; Qianwen Li; Wenyao Xue; Yue Gao; Yu Zeng; Yimeng Wang; Lei Tang; Tiejun Tang; Cong Chen; et al. Gm15575 functions as a ceRNA to up-regulate CCL7 expression through sponging miR-686 in Th17 cells.. null 2020, 125, 32-42, .
- Lei Wu; Jinjin Xia; Donghui Li; Ying Kang; Wei Fang; Peng Huang; Mechanisms of M2 Macrophage-Derived Exosomal Long Non-coding RNA PVT1 in Regulating Th17 Cell Response in Experimental Autoimmune Encephalomyelitisa. Frontiers in Immunology 2020, 11, 1934, 10.3389/fimmu.2020.01934.
- Peijian Yue; Lijun Jing; Xinyu Zhao; Hongcan Zhu; Junfang Teng; Down-regulation of taurine-up-regulated gene 1 attenuates inflammation by sponging miR-9-5p via targeting NF-κB1/p50 in multiple sclerosis. Life Sciences 2019, 233, 116731, 10.1016/j.lfs.2019.116731.
- Chenfan Duan; Yanzhuo Liu; Ying Li; Honglei Chen; Xiaoxiao Liu; Xuewei Chen; Jiang Yue; Xiaoyang Zhou; Jing Yang; Sulfasalazine alters microglia phenotype by competing endogenous RNA effect of miR-136-5p and long non-coding RNA HOTAIR in cuprizone-induced demyelination. Biochemical Pharmacology 2018, 155, 110-123, 10.1016/j.bcp.2018.06.028.
- M. A. Senousy; Olfat G. Shaker; Noha H. Sayed; Nevine Fathy; Mona A. Kortam; LncRNA GAS5 and miR-137 Polymorphisms and Expression are Associated with Multiple Sclerosis Risk: Mechanistic Insights and Potential Clinical Impact. ACS Chemical Neuroscience 2020, 11, 1651-1660, 10.1021/acschemneuro.0c00150.
- Giulia Cardamone; Elvezia Maria Paraboschi; Valeria Rimoldi; Stefano Duga; Giulia Soldà; Rosanna Asselta; The Characterization of GSDMB Splicing and Backsplicing Profiles Identifies Novel Isoforms and a Circular RNA That Are Dysregulated in Multiple Sclerosis. International Journal of Molecular Sciences 2017, 18, 576, 10.3390/ijms18030576.
- Xin Xia; Xinyi Tang; Shengjun Wang; Roles of CircRNAs in Autoimmune Diseases. Frontiers in Immunology 2019, 10, 639, 10.3389/fimmu.2019.00639.
- Leire Iparraguirre; Maider Muñoz-Culla; Iñigo Prada-Luengo; Tamara Castillo-Triviño; Javier Olascoaga; David Otaegui; Circular RNA profiling reveals that circular RNAs from ANXA2 can be used as new biomarkers for multiple sclerosis. Human Molecular Genetics 2017, 26, 3564-3572, 10.1093/hmg/ddx243.
- Jennifer Y. Tan; Keith W. Vance; Miguel A. Varela; Tamara Sirey; Lauren M. Watson; Helen J. Curtis; Martina Marinello; Sandro Alves; Bruno R Steinkraus; Sarah Cooper; et al. Cross-talking noncoding RNAs contribute to cell-specific neurodegeneration in SCA7. Nature Structural & Molecular Biology 2014, 21, 955-961, 10.1038/nsmb.2902.
- Yun Yang; Xinxin Zhou; Yongfeng Jin; ADAR-mediated RNA editing in non-coding RNA sequences. Science China Life Sciences 2013, 56, 944-952, 10.1007/s11427-013-4546-5.
- Maria Anna Zipeto; Qingfei Jiang; Etienne Melese; Catriona H. M. Jamieson; RNA rewriting, recoding, and rewiring in human disease. Trends in Molecular Medicine 2015, 21, 549-559, 10.1016/j.molmed.2015.07.001.
- Ileana Lorenzini; Stephen Moore; Rita Sattler; RNA Editing Deficiency in Neurodegeneration.. Advances in neurobiology 2018, 20, 63-83, 10.1007/978-3-319-89689-2_3.
- Michael S Breen; CommonMind Consortium; Amanda Dobbyn; Qin Li; Panos Roussos; Gabriel Hoffman; Eli Ayumi Stahl; Andrew Chess; Pamela Sklar; Jin Billy Li; et al. Global landscape and genetic regulation of RNA editing in cortical samples from individuals with schizophrenia. Nature Neuroscience 2019, 22, 1402-1412, 10.1038/s41593-019-0463-7.
- Eli Eisenberg; Erez Y. Levanon; A-to-I RNA editing — immune protector and transcriptome diversifier. Nature Reviews Microbiology 2018, 19, 473-490, 10.1038/s41576-018-0006-1.
- Giovanni Enigita; Dario Eveneziano; Alfredo Eferro; A-to-I RNA Editing: Current Knowledge Sources and Computational Approaches with Special Emphasis on Non-Coding RNA Molecules. Frontiers in Bioengineering and Biotechnology 2015, 3, 37-37, 10.3389/fbioe.2015.00037.
- Chammiran Daniel; Jens Lagergren; Marie Öhman; RNA editing of non-coding RNA and its role in gene regulation. Biochimie 2015, 117, 22-27, .
- Galina Shevchenko; Kevin V. Morris; All I's on the RADAR : role of ADAR in gene regulation. FEBS Letters 2018, 592, 2860-2873, 10.1002/1873-3468.13093.
- Giovanni Nigita; Rosario Distefano; Dario Veneziano; Giulia Romano; Mohammad Rahman; Kai Wang; Harvey I. Pass; Carlo M. Croce; Mario Acunzo; Patrick Nana-Sinkam; et al. Tissue and exosomal miRNA editing in Non-Small Cell Lung Cancer. Scientific Reports 2018, 8, 10222, 10.1038/s41598-018-28528-1.
- Takuto Hideyama; Takenari Yamashita; Hitoshi Aizawa; Shoji Tsuji; Akiyoshi Kakita; Hitoshi Takahashi; Shin Kwak; Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiology of Disease 2012, 45, 1121-1128, 10.1016/j.nbd.2011.12.033.
- Hitoshi Aizawa; Takuto Hideyama; Takenari Yamashita; Takashi Kimura; Naoki Suzuki; Masashi Aoki; Shin Kwak; Deficient RNA-editing enzyme ADAR2 in an amyotrophic lateral sclerosis patient with a FUSP525L mutation. Journal of Clinical Neuroscience 2016, 32, 128-129, 10.1016/j.jocn.2015.12.039.
- Takashi Hosaka; Takenari Yamashita; Sayaka Teramoto; Naoki Hirose; Akira Tamaoka; Shin Kwak; ADAR2-dependent A-to-I RNA editing in the extracellular linear and circular RNAs. Neuroscience Research 2019, 147, 48-57, 10.1016/j.neures.2018.11.005.
- Mor Hanan; Alon Simchovitz; Nadav Yayon; Shani Vaknine; Roni Cohen-Fultheim; Miriam Karmon; Nimrod Madrer; Talia Miriam Rohrlich; Moria Maman; Estelle R Bennett; et al. A Parkinson’s disease CircRNAs Resource reveals a link between circSLC8A1 and oxidative stress. EMBO Mol. Med. 2020, 12, e11942, .
- Khen Khermesh; Anna Maria D’Erchia; Michal Barak; Anita Annese; Chaim Wachtel; Erez Y. Levanon; Ernesto Picardi; Eli Eisenberg; Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer's disease. RNA 2015, 22, 290-302, 10.1261/rna.054627.115.
- Minati Singh; Dysregulated A to I RNA editing and non-coding RNAs in neurodegeneration. Frontiers in Genetics 2013, 3, 326, 10.3389/fgene.2012.00326.
- Olivia K Gardner; Lily Wang; Derek Van Booven; Patrice L Whitehead; Kara L Hamilton-Nelson; Larry D Adams; Takiyah D Starks; Natalia K Hofmann; Jeffery M Vance; Michael L Cuccaro; et al. RNA editing alterations in a multi-ethnic Alzheimer disease cohort converge on immune and endocytic molecular pathways. Human Molecular Genetics 2019, 28, 3053-3061, 10.1093/hmg/ddz110.
- Takashi Hosaka; Takenari Yamashita; Akira Tamaoka; Shin Kwak; Extracellular RNAs as Biomarkers of Sporadic Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases.. International Journal of Molecular Sciences 2019, 20, 3148, 10.3390/ijms20133148.
- Yvonne Tay; John L Rinn; Pier Paolo Pandolfi; The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344-352, 10.1038/nature12986.
- Juan Xu; Lin Feng; Zujing Han; Yongsheng Li; Aiwei Wu; Tingting Shao; Na Ding; Lili Li; Wei Deng; Xuebing Di; et al. Extensive ceRNA-ceRNA interaction networks mediated by miRNAs regulate development in multiple rhesus tissues.. Nucleic Acids Research 2016, 44, 9438-9451, 10.1093/nar/gkw587.
- Yan Li; Qiupeng Zheng; Chunyang Bao; Shuyi Li; Weijie Guo; Jiang Zhao; Di Chen; Jianren Gu; Xianghuo He; Shenglin Huang.; et al. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis.. Cell Res 2015, 25, 981-984, .
- Niketa A. Patel; Lauren Daly Moss; Jea-Young Lee; Naoki Tajiri; Sandra Acosta; Charles Hudson; Sajan Parag; Denise R. Cooper; Cesario V. Borlongan; Paula C. Bickford; et al. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. Journal of Neuroinflammation 2018, 15, 1-23, 10.1186/s12974-018-1240-3.
- Shengli Li; Yuchen Li; Bing Chen; Jingjing Zhao; Shulin Yu; Yan Tang; Qiupeng Zheng; Yan Li; Peng Wang; Xianghou He; et al. exoRBase: A database of circRNA, lncRNA and mRNA in human blood exosomes. Nucleic Acids Res 2018, 46, D106-D112, .
- Dewei Wang; Ping Wang; Xianli Bian; Shunliang Xu; Qingbo Zhou; Yuan Zhang; Mao Ding; Min Han; Ling Huang; Jianzhong Bi; et al. Elevated plasma levels of exosomal BACE1‑AS combined with the volume and thickness of the right entorhinal cortex may serve as a biomarker for the detection of Alzheimer's disease. Molecular Medicine Reports 2020, 22, 227-238, 10.3892/mmr.2020.11118.
- Seyedeh Nahid Fotuhi; Mohammad Khalaj-Kondori; Mohammad Ali Hoseinpour Feizi; Mahnaz Talebi; Long Non-coding RNA BACE1-AS May Serve as an Alzheimer’s Disease Blood-Based Biomarker. Journal of Molecular Neuroscience 2019, 69, 351-359, 10.1007/s12031-019-01364-2.
- Brian D. Adams; Christine Parsons; Lisa Walker; Wen Cai Zhang; Frank J. Slack; Targeting noncoding RNAs in disease. Journal of Clinical Investigation 2017, 127, 761-771, 10.1172/jci84424.
- Chun Yao; Bin Yu; Role of Long Noncoding RNAs and Circular RNAs in Nerve Regeneration. Frontiers in Molecular Neuroscience 2019, 12, 165, 10.3389/fnmol.2019.00165.
- Adam P. Carroll; Paul A. Tooney; Murray J. Cairns; Context-specific microRNA function in developmental complexity. Journal of Molecular Cell Biology 2013, 5, 73-84, 10.1093/jmcb/mjt004.
- Christiaan J. Stavast; Stefan J. Erkeland; The Non-Canonical Aspects of MicroRNAs: Many Roads to Gene Regulation. Cells 2019, 8, 1465, 10.3390/cells8111465.
- Juliane C. R. Fernandes; Stephanie M. Acuña; Juliana I. Aoki; Lucile M. Floeter-Winter; Sandra M. Muxel; Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Non-Coding RNA 2019, 5, 17, 10.3390/ncrna5010017.
- Ulf Andersson Ørom; Finn Cilius Nielsen; Anders H. Lund; MicroRNA-10a Binds the 5′UTR of Ribosomal Protein mRNAs and Enhances Their Translation. Molecular Cell 2008, 30, 460-471, 10.1016/j.molcel.2008.05.001.
- Shobha Vasudevan; Yingchun Tong; Joan A. Steitz; Switching from Repression to Activation: MicroRNAs Can Up-Regulate Translation. Science 2007, 318, 1931-1934, 10.1126/science.1149460.
- Min Xiao; Jin Li; Wei Li; Yu Wang; Feizhen Wu; Yanping Xi; Lan Zhang; Chao Ding; Huaibing Luo; Yan Li; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biology 2017, 14, 1326-1334, 10.1080/15476286.2015.1112487.