Safety Pharmacology is one of the most dynamically developing disciplines, whose objective is to assess the potential risks of improperly conducted pharmacotherapy. Evaluation of a potential risks in the use of a given substance is vital for placing a new medicine on the market. The Organisation for Economic Co-operation and Development (OECD) has issued a number of guidelines for drug safety testing [1, 2]. However, the continuous development of medicine demands the extension and refinement of the test panel that a new molecule must undergo before it can be released for use by patients. Toxic effects of compounds on the most important organs are among the most frequent reasons of elimination of substances from further tests. Additionally, safety tests are also performed for medicines already approved for use. Such trials are required, e.g., to register new indications for "old" drugs.
The above mentioned activities require a suitable identifications of the antitargets which are commonly defined as undesirable molecular targets that play an essential role in the proper functioning of cells. Down modulation of an antitarget results in clinically unacceptable side effects, initiation of disease, or deleterious alterations in disease progression. This results in shorter onset time of the disease, increased disease burden, poorer patient outcome, or decreased survival time.
- antitarget
- drug cardiotoxicity
- ion transporter proteins
1. Historical Overview of Drug Cardiotoxicity[1][2]

Figure.1. Drugs withdrawn from market due to proarrhythmic effect (purple), myocardial infraction (green), torsadogenic potential i.e. QT interval prolongation and TdP effect (red).
The statistics show that the risk of cardiotoxicity is one of the most common reasons for withholding or withdrawing drugs from the market[3][4]. This unwanted effect is shared by a large number of non-cardiovascular drug. According to the data up to 70% of potential medicines are either not approved for further testing or their use is significantly limited[5]. Additionally, it has been estimated that approximately 2-3% of all drug prescriptions involve medications that may unintentionally cause long QT syndrome[6]. The first drugs that were removed from clinical use due to cardiotoxicity were encainide (proarrhthmic effect) and terodiline (QT interval prolongation) (Fig.1.)[7][8]. Terdynafine, cisaprid, astemizole, sertindol, thoridazine, grepafloxacin have been removed due to heart toxicity[9] (Fig.1.). In 2020, there was information about QT prolongation for other drugs: osilodrostat, carbetocid, selpercatinib and rucaparib[10]. In 2005 the Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use suggested principles for checking new molecules for the risk of inducing potentially fatal arrhythmias (e.g. torsade the points (TdP)) caused by blocking the human Ether-à-go-go-Related Gene (hERG, KV11.1)[11][12], that was defined to be first antitarget in studies of compound cardiotoxicity[5]. This was based on the observed correlation between electrocardiogram QT prolongation and patients’ risk of TdP arrhythmia. The preclinical and clinical studies proposed in S7B and E14 were implemented, and are now widely used in the pharmaceutical industry and regulatory agencies[11][12]. These assumptions were accepted as the crucial elements of the compound cardiotoxicity assessment. However, further research have demonstrated, that proarrhthmic effect, QT prolongation and hERG blocking cannot be treated as the only determinants of the occurrence of TdP[13]. Verapamil and ralonazine are examples of drugs that are strong inhibitors of the hERG channel and at the same time devoid of the risk of inducing arrhythmias. Moreover, serious disorders of cardiomyocyte electrophysiology was found to be caused by drugs that are weak hERG inhibitors (e.g. sotalol, alluzosin)[14][15]. Thus, it proves the insufficient specificity of the tests based only on the assessment of the hERG channel blocking potential[16]. The risk of drug induced TdP is rather balanced of multiple internal cardiac ionic currents that define ventricular repolarization[17]. Thus, for the first time in 2013, a new strategy was applied for a real proarrhythmic risk assessment of molecules: Comprehensive In Vitro Proarrhythmia Assay (CiPA). CiPA is utilized in the first stages of drug research and focuses on the three most important areas. The first one is the assessment of the interaction of molecules with three ion channel models. In addition to studying the interaction of molecules with hERG, CiPA proposes two additional ion channels gated by voltage, which are essential for the development of arrhythmia: NaV1.5, CaV1.2. The second area of CiPA concern in silico simulations of action potential (AP) responsible for arrhythmia. The third area of CiPA's research is advanced cellular testing using cardiomyocytes derived from pluripotent stem cells (iPSC-CM)[18].
1.1. VGICs - Voltage Gated Ion Channels
The mechanism of opening and closing of ion channels is referred to as gating. It is the progression of the channel through various conformational states. Channel activation is the transition from the rest (closed) state to the open state under the influence of a trigger stimulus (membrane depolarization or ligand binding). Permeation is the passage of ions through the open channel. Therefore, gating process control ion permeation[19] participating in cellular signaling is dependent on cations flow driven by electrochemical gradient. Voltage-gated Na+, Ca2+, and K+ channels share a common molecular architecture. They also possess the same set of three voltage-dependent functionally distinct states i.e. closed (or resting), activated (or open), and inactive[6]. Under physiological conditions, the Na+ and Ca2+ channels are inward cell currents and those flowing through the K+ channels are outward currents. Besides the channels that open or close depending on the value of the cell transmembrane potential (so-called Voltage-Gated Ion Channels (VGICs)) there are also channels gating independently on transmembrane potential, controlled by other external or intracellular factors.
According to the research of International Union of Primary and Clinical Pharmacology (IUPHAR) and the British Pharmacological Society (BPS), ion channels are the third largest target group for drugs[20][21]. They participate in controlling physiological and pathological processes in the body. VGICs take part in generating and transmitting information within the cells of the central and peripheral nervous and cardiovascular systems[22]. It should be emphasized that Kv11.1, Nav1.5 and Cav1.2 ion channels, not only represent useful targets with potential therapeutic applications, but also indicate undesirable targets (also known as antitargets) to be avoided because of the side effects they cause when their function is altered[23].
1.2. VGICs - Structure
The molecular architecture of the ion channel families consists of four variations built on a common pore forming structural theme. Ion channels consist of poreforming a-subunits (also called a1-subunits) and accessory a2-, b-, d-, g-subunits. The a-subunit is inserted into the lipid bilayer of the cellular membrane and constitutes the channel pore, through which ions pass (Fig. 2). Commonly, a-subunits and accessory subunits are members of large protein families that evolutionary possess similar structural elements i.e. comparable amino acid sequences. This is reflected in the names of the subunits and their genes. For example, the gene encoding the a-subunit of the cardiac Ca2+ channel is called CACNC1C: calcium channel, isoform 1, a-subunit. The a-subunit is termed CaV1.2: Ca2+ channel family, subfamily 1, member 2; the subscript “V” means that channel gating is regulated by transmembrane voltage changes (voltage dependent)[24].
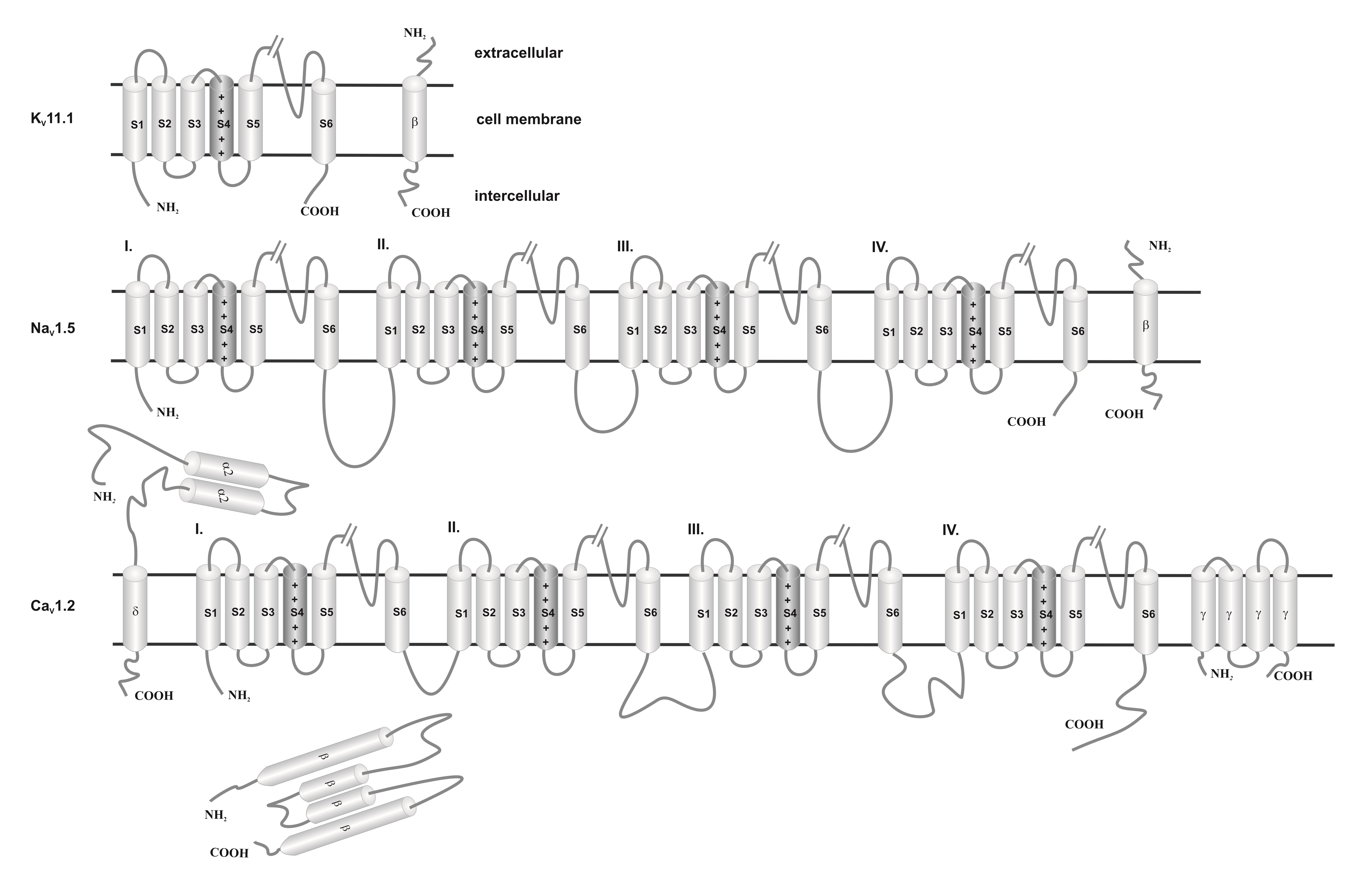
Figure. 2. Schematic view of the structure’s subunits of cardiac ion channels. a1-subunits of K+ consist of one single subunit (domain or core motif), while Na+ and Ca2+ consist of four serially linked homologous domains (I-IV). Each subunit containing six transmembrane segments (S1-S6). The S5 and S6 segments and the membrane-associated pore loop (often called the P loop or P segment or P region) between them form the central pore through which ions flow down their electrochemical gradient. The S4 transmembrane domain is the voltage-sensor (gating modifier). K+ channel is an assembly of subunits with a single core motif. While Na+ and Ca2+ channels consist of four subunits co-assembled to form a single functional channel. The b, a2d and g subunits enhance cell surface expression and modulate the voltage dependence and gating kinetics of the a1-subunits and channel sensitivity to endogenous ligands and pharmacological agents. These accessory subunits determine ion channel tissue specificity.
Table 1. Overview of the key properties of the structural subunits of cardiac ion channels[25].
|
name |
hKv11.1/KCNH2 |
hNav1.5/SCN5A |
hCav1.2/CACNC1C |
|
UniProtKB |
Q12809 |
Q14524 |
Q13936 |
|
pore-forming |
612-632: VTALYFTFSSLTSVGFGNVSP |
884-904: FFHAFLIIFRILCGEWIETMW |
694-715: QSLLTVFQILTGEDWNSVMYDG |
|
ion selectivity sequence motif |
GYG |
DEKA |
EEEE |
|
cariac disease |
Long Qt Syndrome; Short Qt Syndrome |
Atrial Fibrillation, Familial, Brugada Syndrome; Cardiomyopathy, Dilated; Long Qt Syndrome; Progressive Familial Heart Block (Type Ia); Sick Sinus Syndrome, Autosomal Recessive; Sudden Infant Death Syndrome; Ventricular Fibrillation During Myocardial Infarction, Susceptibility to acquired arrhythmia |
Long QT, Brugda Syndrome, Timothy Syndrome |
|
sequence identity* |
88.66% |
66.70% |
70.3% |
*Sequence identity between the template and the modeled sequence
2. Mechanism of Ion Channel Inhibition
Although the general mechanism of ion channel inhibition is well known, the detailed description is still unclear and controversial. Voltage-dependent gating can be triggered in variety of ways, and the mechanisms of VGIC operation are important tool to understand the signaling behavior of the channel[26]. The mechanisms of ion channel inhibition can be categorized in two classes, i.e., pore plugging, and allosteric binding. The former includes inhibitors capable of binding in the pore region once they enter the channel, in consequence they physically block the pore disabling the ion transport. The latter mechanism involves inhibitors that require a specific binding site, the site usually is extracellular side of the pore, but there are known exceptions. The allosteric inhibitor binds to the channel at the binding site causing conformational changes of the protein that prevent the normal function of the channel[6]. Table 1 summarizes the pore forming region in Kv11.1, Nav1.5 and Cav1.2 channels.
3. In Silico Methods for Testing the Tisk of Cardiotoxicity
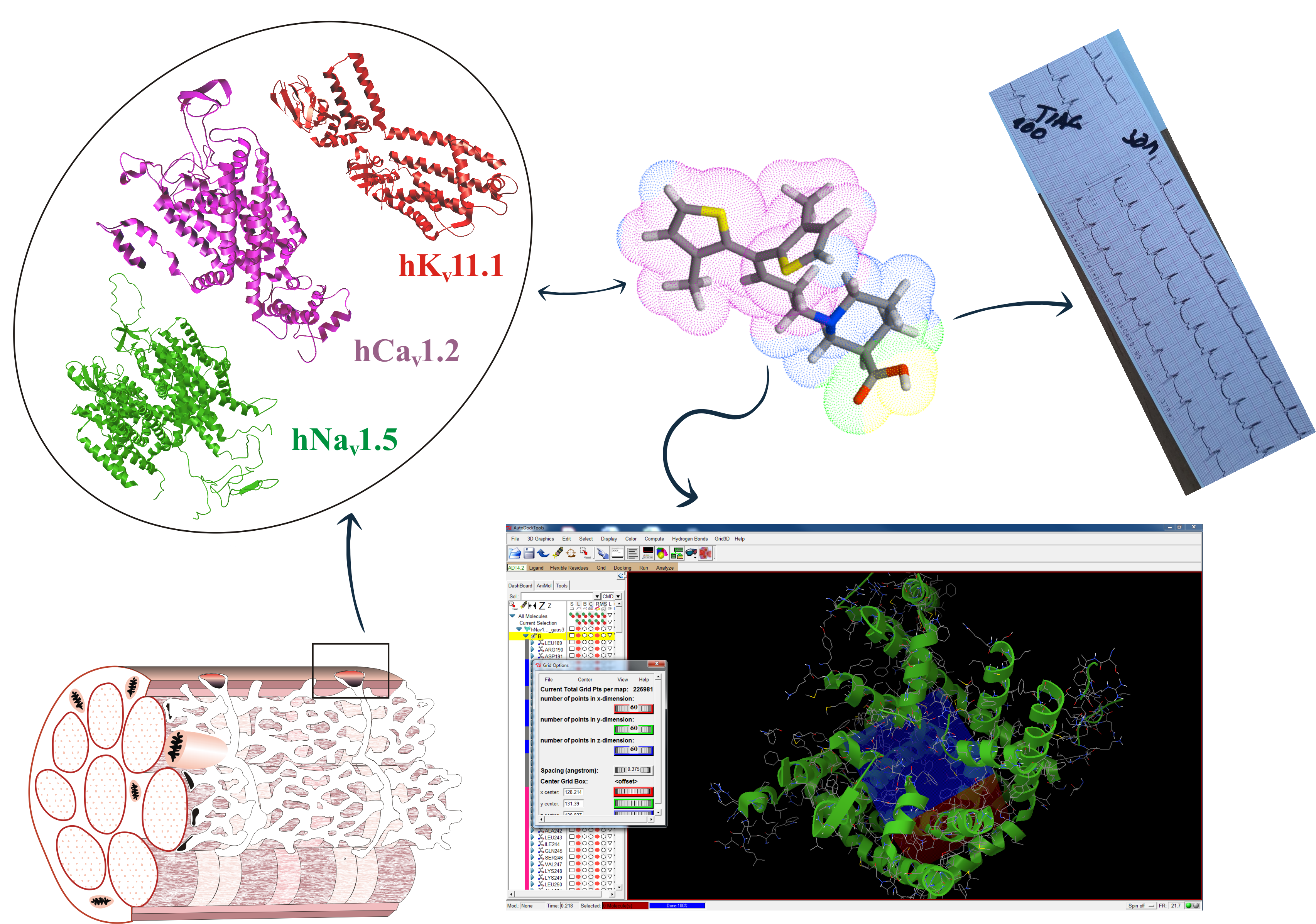
Figure. 3. Scheme of the calculation procedure.
One of the most popular and accurate in silico methods is the molecular docking technique[27]. This method determines the affinity (the binding of compounds to the channel) and how a given drug binds to the active site of a protein (also known as binding modes/pose). It is a source of information about the physiological and pathological mechanism of action of many substances[28]. It should be mentioned here that channels KV11.1, NaV1.5, CaV1.2 can be treated in two ways. First, they are therapeutic targets for cardiac arrhythmia and hypertension. Secondly, they should be treated as antitargets in cardiac risk assessment. This entails the necessity of opposite approaches in interpretation of the channels blocking effects. In the first case, research is conducted to search for molecules that bind to a specific therapeutic target (e.g. Class III antiarrhythmic drugs). In the second, research is focuses on looking for molecules that avoid strong binding to the unwanted targets (i.e. many noncardiac drugs)[5][29]. Commonly, Ki is characterized by the dissociation constant of the complex, which is determined by competitive radioligand binding, and the blocking of the passage of ion through the channel. In the computational approach, Ki is determined in the computational process. In vivo desirable and undesirable effects of a drug are generally related to its concentration at the sites of action[30]. The passage of some ions through the channel is characterized by the IC50 concentration, ensuring a 50% reduction in selected ion current, which is determined by electrophysiological methods in the channel-expressing cells. For majority of compounds, only data obtained by one of experimental methods are available. It should be emphasized here that the values of these parameters are generally well correlated (IC50 ≈ Ki). Based on the assumption that the calculated Ki reflects the drug binding affinity for a specific therapeutic target. On the other hand, the IC50 (or pIC50) more reflects the functional potency of the inhibitor for the drug. It is widely accepted that the use of calculated Ki (or pKi) is helpful in determining the likelihood that a given drug will inhibit a particular protein target and result in a clinically relevant drug interaction. For this we usually assume that the IC50 corresponds to the numerical value of Ki (or pIC50 ≈ pKi). By convention, a pIC50 ≤ 4 (or pKi) is commonly used as threshold[31][32]. In the publication of M. Kowalska et al. pIC50 of selected drugs and their corresponding ion channels are summarized in Table 2[33]. By matching the calculated Ki values with the corresponding tabular data, we are able to assess, for example, the cardiotoxicity of drugs by analyzing their interaction with ion channels[34]. From safety pharmacological point of view, lower pIC50 (pKi) value for channels Kv11.1, Nav1.5, Cav1.2 are desirable.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21218099
References
- OECD Test No. 453: Combined Chronic Toxicity/Carcinogenicity Studies, OECD Guidelines for the Testing of Chemicals, Section 4.
- OECD, T. G. G., The Organisation for Economic Co-operation and Development. OEEC: 1960.
- Kirsch, G. E.; Kramer, J.; Bruening-Wright, A.; Obejero-Paz, C.; Brown, A. M., The Comprehensive In Vitro Proarrhythmia Assay (CiPA) Guide: A New Approach to Cardiac Risk Assessment. 2016.
- Li, Z.; Ridder, B. J.; Han, X.; Wu, W. W.; Sheng, J.; Tran, P. N.; Wu, M.; Randolph, A.; Johnstone, R. H.; Mirams, G. R., Assessment of an In Silico Mechanistic Model for Proarrhythmia Risk Prediction Under the Ci PA Initiative. Clinical Pharmacology & Therapeutics 2019, 105, (2), 466-475.
- Raschi, E.; Vasina, V.; Poluzzi, E.; De Ponti, F., The hERG K+ channel: target and antitarget strategies in drug development. Pharmacological research 2008, 57, (3), 181-195.
- Petkov, G. V., Ion channels. In Pharmacology, Elsevier: 2009; pp 387-427.
- Cheung, S.; Parkinson, J.; Wählby-Hamrén, U.; Dota, C.; Kragh, Å.; Bergenholm, L.; Vik, T.; Collins, T.; Arfvidsson, C.; Pollard, C., A tutorial on model informed approaches to cardiovascular safety with focus on cardiac repolarisation. Journal of pharmacokinetics and pharmacodynamics 2018, 45, (3), 365-381.
- Sallam, K.; Li, Y.; Sager, P. T.; Houser, S. R.; Wu, J. C., Finding the rhythm of sudden cardiac death: new opportunities using induced pluripotent stem cell–derived cardiomyocytes. Circulation research 2015, 116, (12), 1989-2004.
- Lee Hyang-Mi; Yu Myeong-Sang; Kazmi Sayada Reemsha; Oh Seong Yun; Rhee Ki-Hyeong; Bae Myung-Ae; Lee Byung Ho; Shin Dae-Seop; Oh Kwang-Seok; Ceong Hyithaek; Lee Donghyun; Dokyun, N., Computational determination of hERGrelated cardiotoxicity of drug candidates. BMC Bioinformatics 2019, 20, 67-73.
- Woosley, R. L.; Heise, C. W.; Romero, K. A.
- Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. 2005.
- Use, I. C. f. H. o. T. R. f. P. f. H., ICH E14 Guideline: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs. 2015.
- Li, Z.; Dutta, S.; Sheng, J.; Tran, P. N.; Wu, W.; Chang, K.; Mdluli, T.; Strauss, D. G.; Colatsky, T., Improving the in silico assessment of proarrhythmia risk by combining hERG (human ether-à-go-go-related gene) channel–drug binding kinetics and multichannel pharmacology. Circulation: Arrhythmia and Electrophysiology 2017, 10, (2), e004628.
- Hondeghem, L.; Carlsson, L.; Duker, G., Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation 2001, 103, (15), 2004-2013.
- Mirams, G. R.; Cui, Y.; Sher, A.; Fink, M.; Cooper, J.; Heath, B. M.; McMahon, N. C.; Gavaghan, D. J.; Noble, D., Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovascular research 2011, 91, (1), 53-61.
- Martin, R. L.; McDermott, J. S.; Salmen, H. J.; Palmatier, J.; Cox, B. F.; Gintant, G. A., The utility of hERG and repolarization assays in evaluating delayed cardiac repolarization: influence of multi-channel block. Journal of cardiovascular pharmacology 2004, 43, (3), 369-379.
- Blinova Ksenia; Stohlman Jayna; Vicente Jose; Chan Dulciana; Johannesen Lars; HortigonVinagre Maria P.; Zamora Victor; Smith Godfrey; Crumb William J.; Pang Li; Lyn-CookBeverly; Ross James; Brock Mathew; Chvata Stacie; Millard Daniel; Galeotti Loriano; Stockbridge Norman; G., S. D., Comprehensive Translational Assessment of Human Induced Pluripotent Stem Cell Derived Cardiomyocytes for Evaluating Drug-Induced Arrhythmias. Toxicol Sci 2017, 155, (1), 234-247.
- Sager Philip T.; Gintant Gary; Turner Rick; Pettit Syril; Norman, S., Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am Heart J 2014, 167, (3), 292-300.
- Gingrich, K. J.; Yang, J., Molecular physiology. Foundations of Anesthesia: Basic Sciences for Clinical Practice 2006, 79.
- Dunlop J.; Peri R.; Vasilyev D.; R., A., High-throughput electrophysiology: an emerging paradigm for ion channel screening and physiology. Nat Rev Drug Discov 2008, 7, 358-368.
- Harmar Anthony J.; Rosser Edward M. ; Jones Martin; Buneman Peter; Dunbar Donald R. ; Greenhill Stuart D.; Hale Valerie A. ; SharmanJoanna L. ; Bonner Tom I.; Catterall William A.; Davenport Anthony P.; Delagrange Philippe; Dollery Colin T.; Foord Steven M.; Gutman George A.; Laudet Vincent; Neubig Richard R.; Ohlstein Eliot H.; Olsen Richard W.; Peters John; Pin Jean-Philippe; Ruffolo Robert R.; Searls David B.; Wright Mathew W.; Michael., S., IUPHAR-DB: the IUPHAR database of G protein-coupled receptors and ion channels. Nucleic Acids Research 2008, 37, 81-86.
- Barker B.S.; Young G.T.; Soubrane C.H.; Stephens G.J.; Stevens E.B.; M.K., P., Ion Channels: chapter 2. 2017; p 11-43.
- Fernández-Ballester , G.; Fernández-Carvajal, A.; González-Ros, J. M.; Ferrer-Montiel, A., Ionic Channels as targets for drug design: a review on computational methods Pharmaceutics 2011, 3, 932-953.
- Amin, A. S.; Tan, H. L.; Wilde, A. A., Cardiac ion channels in health and disease. Heart Rhythm 2010, 7, (1), 117-126.
- Alexander, S. P.; Cidlowski, J. A.; Kelly, E.; Mathie, A.; Peters, J. A.; Veale, E. L.; Armstrong, J. F.; Faccenda, E.; Harding, S. D.; Pawson, A. J., The Concise Guide to PHARMACOLOGY 2019/20: Ion channels. British journal of pharmacology 2019, 176, S229-S246.
- Yellen, G., The moving parts of voltage-gated ion channels. Quarterly reviews of biophysics 1998, 31, (3), 239-295.
- Chang, C.; Ray, A.; Swaan, P., In silico strategies for modeling membrane transporter function. Drug discovery today 2005, 10, (9), 663-671.
- Radchenko, E.; Rulev, Y. A.; Safanyaev, A. Y.; Palyulin, V.; Zefirov, N. In Computer-aided estimation of the hERG-mediated cardiotoxicity risk of potential drug components, Doklady Biochemistry and Biophysics, 2017; Springer: 2017; pp 128-131.
- Recanatini, M.; Poluzzi, E.; Masetti, M.; Cavalli, A.; De Ponti, F., QT prolongation through hERG K+ channel blockade: current knowledge and strategies for the early prediction during drug development. Medicinal research reviews 2005, 25, (2), 133-166.
- Huang, S. M.; Temple, R.; Throckmorton, D.; Lesko, L., Drug interaction studies: study design, data analysis, and implications for dosing and labeling. Clinical Pharmacology & Therapeutics 2007, 81, (2), 298-304.
- Zaręba, P.; Gryzło, B.; Malawska, K.; Sałat, K.; Höfner, G. C.; Nowaczyk, A.; Fijałowski, Ł.; Rapacz, A.; Podkowa, A.; Furgała, A.; Żmudzki, P.; Wanner, K. T.; Malawska, B.; Kulig, K., Novel GABA uptake inhibitors with enhanced inhibitory activity toward mGAT3/4 and their effect on pain threshold in mice. EJMCh 2020, 188, (111920).
- Böck, M. C.; Höfner, G.; Wanner, K. T., N‐Substituted Nipecotic Acids as (S)‐SNAP‐5114 Analogues with Modified Lipophilic Domains. ChemMedChem 2020.
- Kowalska, M.; Nowaczyk, J.; Nowaczyk, A., KV11.1, NaV1.5, and CaV1.2 transporter proteins as antitarget for drug cardiotoxicity. Int. J. Mol. Sci. 2020, 21, 8099.
- Nowaczyk, A.; Fijałkowski, Ł.; Zaręba, P.; Sałat, K., Selective neuronal and astrocytic inhibition of human GABA transporter isoform 1 (hGAT1) inhibitors in the mechanism of epilepsy and pain - molecular docking and pharmacodynamics studies, part I. Journal of Molecular Graphics and Modelling 2018, 85, 171-181.