Tuberculosis (TB) is still the world’s leading infectious cause of death, according to the World Health Organization (WHO) [
1,
2]. Its etiological agent,
Mycobacterium tuberculosis (
Mtb), kills approximately two million people every year and latently infects one third of the world’s population. Latency is one of the most remarkable features of TB infection, where
Mtb establishes a dynamic equilibrium with the host immune system that lasts for lifetime, with no signs or symptoms of disease [
3,
4]. It is assumed that during latency,
Mtb persists in host tissues mostly in a dormant state. Resuscitation from dormancy, which is orchestrated by a set of cell wall hydrolases [
5,
6,
7,
8,
9], is a regular event in the homeostasis of
Mtb infection that continuously replenishes the bulk of replicating bacilli after their elimination by the host immune response [
10,
11]. TB reactivation occurs when the equilibrium between
Mtb and the host immune response is broken in favor of bacterial replication and tissue damage.
Active TB disease is curable with long-lasting multidrug therapeutic regimens, but the emergence of drug-resistant TB represents a major obstacle to future TB care, with important economic and social consequences [
12]. Ambitious goals for better controlling the global TB epidemic can be met with the development of new drug treatments, improved diagnostics, and most importantly the availability of a new and more effective vaccine [
1,
2]. At present, and 100 years after its introduction,
Mycobacterium bovis Bacille Calmette and Guérin (BCG) is the only vaccine available for TB control [
13,
14], though its protective activity in preventing TB in adults is variable, incomplete, and overall insufficient [
15,
16,
17]. There is an urgent need for a new and more effective vaccine, yet poor understanding of the complex relationship between
Mtb and the human immune system, paired with the lack of immunological correlates of protection, makes this endeavor challenging [
18].
Knowing the three-dimensional structure of an antigen provides important insights into the understanding of the molecular nature of host–pathogen interactions and of the key epitopes that may serve as a target for the host antibody response. Although structural data on PE_PGRS proteins are not available, insightful information can be obtained by modeling techniques, learning from homologous proteins. Among PE_PGRS proteins, PE_PGRS33 is one of the best studied for its interaction with the immune system and has been considered a model for PE_PGRSs. As such, its putative role as a vaccine candidate is worth being investigated [
81]. PE_PGRS33 is a large protein of 498 residues, with a modular architecture. A search in the PFAM database only identifies a PE domain at the N-terminal region of the protein, a small domain (residues 1–93) that is distributed nearly exclusively in actinobacteria, with only one exception for
Tulasnella calospora, a genus of patch-forming fungi in the
Tulasnellaceae family. A conserved linker GRLPI domain (l-GRPLI), likely acting as a transmembrane anchor, connects the PE domain to a distinctive region, not predicted by PFAM, and characterized by multiple repeats containing the GGA-GGX motif interspersed with unique sequences, and commonly denoted as PGRS domain ().
2.1. The PE Domain
The PE domain takes its name from the conserved Pro–Glu (PE) amino acids at its N-terminus (residues 7–8) [
55] ). This domain is responsible for PE_PGRS33 translocation via ESX5 and cell wall localization, with a significant role of 30 amino acids on its N-terminus [
82]. A search in the PFAM database shows that the PE family is a member of clan EsxAB (CL0352), which also includes the more common PPE family and the WXG100 family, including the well-known antigen ESAT-6 (6 kDa early secreted antigenic target) and CFP-10 (10 kDa culture filtrate protein) in
Mtb or EsxA (ESAT-6-like extracellularly secreted protein A) and EsxB in
Staphylococcus aureus.
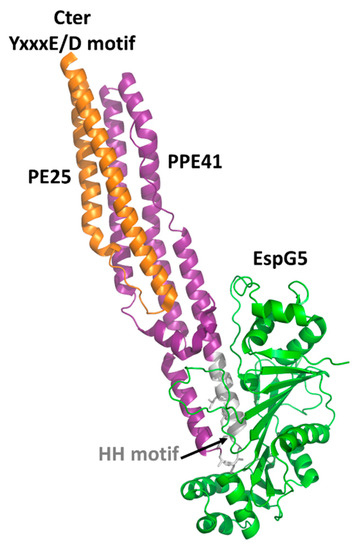
Crystal structures have been reported for the two PE domains PE8 and PE25, in both cases forming a heterocomplex with PPE partners (). In all structures, PE and PPE interact via a hydrophobic interface forming a four-helix bundle formed by two α-helices from the PE and two α-helices from the PPE module (). The crystal structure of the ESX-5-secreted PE25–PPE41 heterodimer in complex with the ESX-5-encoded cytoplasmic chaperone EspG5 shows that EspG5 binds to a highly conserved hydrophobic chaperone-binding sequence on PPE, named as the hh motif [
83] ().
Figure 2. Cartoon representation of the crystal structure of PE25-PPE41 (orange/purple) ternary complex with the chaperone EspG5 (green). The signature ESX secretory motif YxxxE/D is located at the C-terminal side of PE25 (orange), whereas the EspG5 binding region is located on the HH motif of PPE41 (gray) [
83].
Table 2. Structures of PE-like domains in Mycobacterium tuberculosis (Mtb).
By binding PPE, EspG5 protects the aggregation-prone hh motif on PPE proteins and keeps the dimers in a secretion-competent state. Consistently, point mutations of this conserved hh motif affect protein secretion [
83]. Both in the cases of PE25–PPE41 and PE8–PPE15, the binding of EspG5 chaperone does not cause conformational changes in the heterodimers. The two ternary complexes present highly similar structures. A superposition of their structures using DALI produces root mean square deviations (rmsd), computed on the backbone atoms of PE, PPE, and EspG5 chains, of 1.1 Å, 2.1 Å, and 0.5 Å, respectively. Importantly, EspG5 binds the PE–PPE dimers at a location that does not interfere with the signature ESX secretory motif YxxxD/E at the C-terminal side of PE proteins [
87] ().
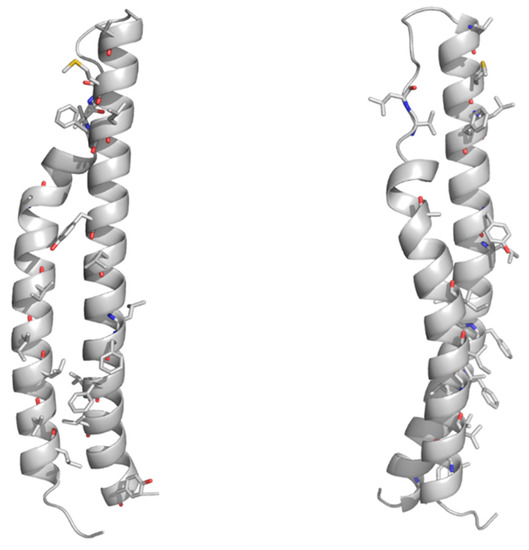
As mentioned above, no structural information on the PE domains from PE_PGRS proteins is hitherto known. Neither it is known whether PE_PGRS proteins strictly require a PPE-like domain, as in the case of PE proteins in . Here, we fill this structural gap by adopting homology modeling. The best template was identified by HHPRED as the PE domain the ESX-5-secreted PE8 (PDB code 5xsf, sequence identity 45.8%) and the homology model built using MODELLER [
88,
89,
90,
91]. As a result, the homology model of the PE domain of PE_PGRS33 shows that all hydrophobic/aromatic residues are located on one side of the molecule (). This feature, also observed for the PE domains of PE/PPE complexes, suggests that either the PE domain of PE_PGRS33 forms homodimers or it is prone to interact with another protein to form a heterodimer. It is hitherto not clear whether PE_PGRS proteins require a protein partner [
59,
86], as in the case of PE/PPE proteins. Indeed, pe_pgrs genes are expressed as single operons. Also, the PE-unique LipY protein does not require a partner to be secreted [
92]. These findings suggest that PE_PGRSs can be stable on their own, albeit being endowed with prone-to-interact PE domains for their functions.
Figure 3. Cartoon representations of the homology model of the PE domain of PE_PGRS33. Front and side views are reported on left and right sides, respectively. The model was computed with MODELLER using the structure of the PE25 domain from a type VII secretion system of Mycobacterium tuberculosis (Mtb) as a template (PDB core 4w4k, sequence identity 37%). Hydrophobic and aromatic residues are drawn in stick representation.
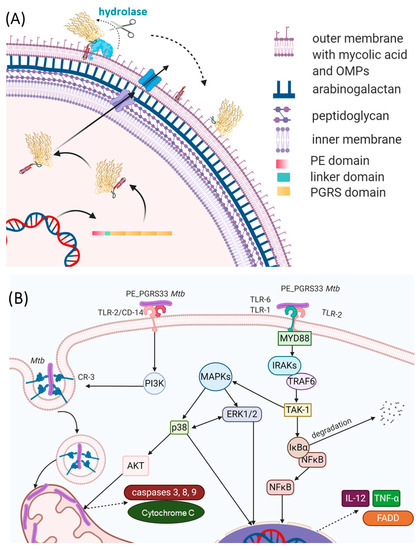
Importantly, the PE domain of PE_PGRS33 is required for the protein translocation through the mycobacterial cell wall [
63,
82,
88,
90,
91]. Once exerted this role, the PE domain is cleaved from the rest of the molecule, leaving the functional PGRS domain floating on the mycomembrane [
59]. Therefore, it is tempting to surmise that some hydrolases may recognize PE_PGRS33 through its PE domain.
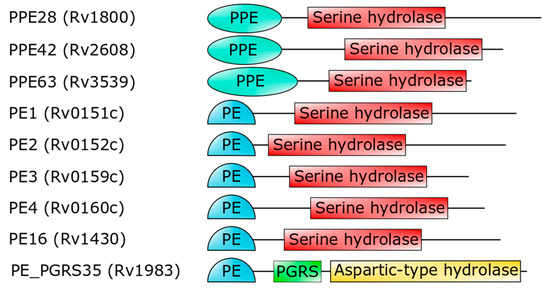
Mtb encodes for a number of PE- and PPE-containing serine α/β hydrolases, which are possible candidates as PE_PGRS33 hydrolases [
93] (). In addition to these, a PE_PGRS aspartic-type endopeptidase, denoted as PecA in
M. marinum and PE_PGRS35 in
Mtb, is known to cleave the lipase LipY [
92]. More investigations are needed to verify which hydrolase is responsible for PE cleavage of PE_PGRS33 and if more hydrolases cleave specific PE_PGRS proteins.
Figure 4. Domain architecture of PE- or PPE-containing
Mtb proteins (strain H37Rv), which are predicted to embed a serine hydrolase domain [
93]. The last hydrolase, PE_PGRS35, is the
Mtb homolog of
M. marinum PecA [
92].
2.2. PGRS Domain Contains Multiple PGII Modules
In PE_PGRS proteins, the PGRS domain can vary in size from tens to almost 1800 amino acid residues. Its main feature is the presence of multiple repeats containing the GGA-GGX motif interspersed with unique sequences [
94]. It has been shown that PGRS domains are available on the mycobacterial surface and can directly interact with host components, as TLR2 receptors [
60,
74,
95]. To date, the structure of the PGRS domain remains unknown and should be implemented in experimental data.
The lack of structural data on PGRS domains makes the understanding of the role of these domains a hard task. However, a high sequence identity of the C-terminal part of the PGRS domain of PE_PGRS33 exists with the PG
II domain of snow flea antifreeze protein sfAFP from
Hypogastrura harveyi (sequence identity 60% with residues 406–486). Therefore, we performed homology modeling based on the target–template alignment using ProMod3 and the structure of sfAFP as a template (PDB code 2pne). The alignment of the sequence of this C-terminal PG
II module against the entire PGRS region identifies further three modules with the same pattern and sequence identities ranging between 63.0% and 53.9% (
Figure S1). This analysis shows that the PGRS domain of PE_PGRS33 is formed by four PG
II domains, denoted here as PG
II1, PG
II2, PG
II3, and PG
II4, all with similar structural features.
Polyglycine conformations, such as PG
II, are the most flexible ones because the lack of side chains in glycine removes steric hindrances. Consequently, extended regions of the Ramachandran plot are allowed for glycine residues, which can virtually assume any
ψ angle [
96]. Typical of PG
II conformation, each glycine-rich triplet folds into a left-handed, elongated helix with a pitch (rise per turn) of 9.2 Å. This conformation resembles that observed for the polyproline type II (PP
II) helices found in collagen [
97,
98]. In the PG
II sandwich, six antiparallel PG
II helices are stacked in two antiparallel groups, with three to four triplets spanning the PG
II domain length (). The organization of the PGRS region in PG
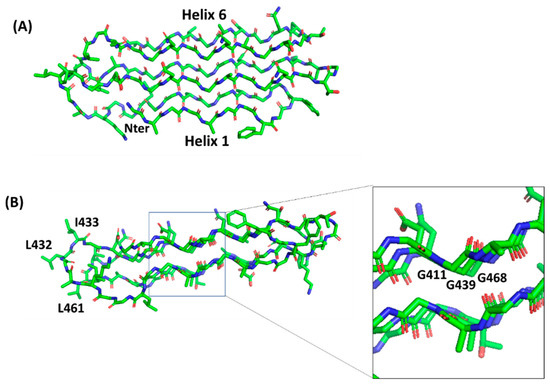
II domains explains the high abundance of glycine residues in these domains. Glycine residues are always pointing inwards, in positions where only glycine could be sterically allowed ().
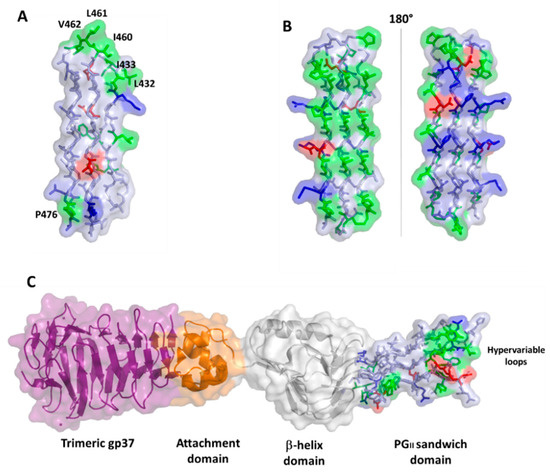
Figure 5. Stick representation of the homology model of the PGII sandwich domain PGII4, computed using the structure of sfAFP as a template (pdb code 2pne). The front view (A) shows the six PGII helices, whereas the side view (B) shows the localization of hydrophobic residues (e.g., L432, I433, L461) on the lateral loops. The inset shows glycine residues pointing inside and stabilizing the tightly packed PGII helices.
A comparison with the structure of the PE_PGRS33 PG
II domains with that of the antifreeze protein sfAFP highlights different surface characteristics, albeit presenting the same fold, likely due to completely different functions of PG
II domains in the two proteins (). In PE_PGRS33, hydrophobic residues of PG
II domains are located mainly on loop regions (B and A), likely accounting for a role of these residues in host recognition. Consistently, as will be shown later, removal of consecutive residues belonging to PG
II domains of PE_PGRS33 (alleles from 48 to 52, ) results in weaker immunostimulatory activity, in terms of reduced TNF-α [
72,
74]. By contrast, hydrophobic residues are mostly located on one side of the PG
II structure of sfAFP (B) [
99]. The accumulation of hydrophobic residues on one side of the sandwich builds a module with one hydrophilic face and one hydrophobic face, and the flat hydrophobic face is supposed to interact tightly with the highly ordered water molecules found at the surface of an ice crystal [
99]. In this respect, the flatness that characterizes this domain is functional for its tight association with the ice surface [
99]. Interestingly, a PG
II sandwich was also observed in the
Salmonella bacteriophage S16 long tail fiber. In this case, this PG
II sandwich domain plays a role in the interactions of the phage with its host, with its exposed (hydrophobic) loops being determinants of host binding. Therefore, similar to PG
II of PE_PGRS33, the glycine-rich core of the PG
II sandwich of S16 long tail fiber exposes hypervariable β-turn loops that determine receptor specificity (C).
Figure 6. Surface and stick representations of PGII domains in (A) PE_PGRS33 (domain PGII4); (B) antifreeze protein sfAFP (pdb code 2pne) in two 180° views; and (C) Salmonella bacteriophage S16 long tail fiber (pdb code 6F45). In this panel, the PGII domain is located at the C-terminal side of the protein (stick and surface representation). Adjacent domains are drawn in surface and cartoon representations. In all panels, the color code used for PGII residues is red for negative, blue for positive, green for hydrophobic, and light blue for polar residues.
In contrast to the antifreeze protein sfAFP, it is likely that in the case of both PE_PGRS33 and the bacteriophage S16 long tail fiber, the flatness of the PG
II sandwich is useful to allow recognition loops to be closely spaced. In both cases, these loops evolve rapidly, as confirmed by their hypervariable nature, in a similar manner as observed for the three hypervariable complementarity-determining regions (CDRs) of immunoglobulins [
100,
101]. As in the case of S16 tail fiber, the PG
II loops of PE_PGRS33 are likely exposed to the host and, as such, the principal targets of antibodies.