The molecular mechanisms controlling glycolytic reprogramming upon TLR stimulation have been partially uncovered and differ according to cell types. In murine BMDCs stimulated through TLR4 by LPS, the early increase in glycolysis is controlled by activation of TBK1, IKKε and AKT kinases, favoring mitochondrial translocation of HK2 [
9] (). Relocalization of HK2 to the mitochondria, occurring within the first hour of LPS stimulation, enhances HK activity, thus participating in the early glycolytic burst, fueling the TCA cycle and fatty acids (FA) synthesis. Glycogen metabolism supports early glycolytic reprogramming required for DC immune responses [
13, 14]. The modulation of pyruvate kinase M2 (PKM2) activity, another key enzyme of glycolysis, is required to induce inflammatory responses in macrophages. Deacetylation of PKM2 by class II histone deacetylases enhances its activity and thus promotes LPS-inducible interleukin (IL)-1β production in human and mouse macrophages [
15]. Upon TLR4 stimulation, the expression of inducible nitric oxide synthase (iNOS) is also upregulated in mouse macrophages and DCs. This enzyme catalyzes the production of nitrogen oxide (NO) from L-arginine. NO, that diffuses in the microenvironment, then interferes with TCA cycle functioning and inhibits mitochondrial electron transport chain complexes, thus reducing O
2 consumption and ATP production by OXPHOS [
32]. The late increase in glycolytic metabolism observed in these cells may be a survival mechanism to maintain ATP production despite OXPHOS inhibition [
4]. Importantly, human macrophages, which do not produce significant NO, present unaltered mitochondrial respiration upon LPS stimulation [
58,
59].
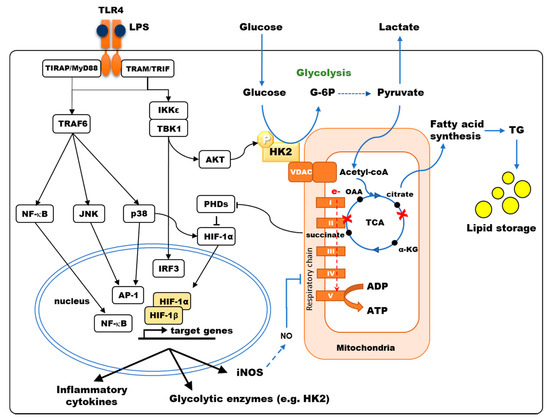
Figure 1. TLR4 signaling modulates the central carbon metabolism of macrophages and myeloid dendritic cells. TLR4 stimulation results in the activation of NF-κB, JNK, p38-MAPK, inducing the secretion of pro-inflammatory cytokines, and of TBK1/IKKε inducing IRF3-dependent type I interferon and AKT phosphorylation, favoring Hexokinase-2 (HK2) binding to VDAC. Hexokinase (HK) activity is the rate-limiting enzyme controlling glucose entry into glycolysis. Pyruvate is converted to acetyl-CoA into the mitochondria, fueling the TCA cycle. LPS stimulation of murine bone-marrow-derived DCs and macrophages results in a broken TCA cycle (red crosses), where succinate accumulates, inhibiting prolyl-hydroxylase domain enzymes (PHDs) thus favoring HIF-1α accumulation, and citrate is diverted from the TCA cycle to fuel fatty acid synthesis. HIF-1α induces the transcription of genes such as glycolytic enzymes, inducible nitric oxide synthase (iNOS) and pro-IL-1β. In human monocyte-derived DCs, p38-MAPK activation results in HIF-1α accumulation, enhancing the expression of metabolic enzymes such as HK2. Under aerobic conditions, electron (e-) transport through the respiratory chain (Complex I to V) generates ATP by oxidative phosphorylation (OXPHOS). In murine DCs, OXPHOS is inhibited by NO production upon LPS stimulation.
Hypoxia-inducible factor 1-alpha (HIF-1α) is a master transcriptional regulator of glycolytic enzymes, including HK and PKM2 [
60]. Under normoxia, HIF-1α degradation is induced by prolyl-hydroxylase domain enzymes (PHDs) that hydroxylate proline and asparagine residues. The interaction of the von Hippel–Lindau (VHL) factor with hydroxylated residues of HIF-1α recruits another component of an E3 ubiquitin–ligase that targets this factor to the proteasome for degradation [
29]. PHD requires O
2 and α-ketoglutarate (α-KG) to hydroxylate HIF-1α and generates CO
2 and succinate as byproducts of this reaction. Hence, PHD activity is controlled by intracellular concentrations of O
2 and α-KG as substrates but is also regulated by succinate, which is acting as a competitive inhibitor of these enzymes. Hypoxia inhibits PHDs, resulting in HIF-1α accumulation [
61]. Nevertheless, in normoxic conditions, alternative mechanisms also result in HIF-1α accumulation upon LPS stimulation. PHD inhibition depends on reactive oxygen species (ROS) production and succinate accumulation, thus increasing HIF-1α stability [
57,
62]. In murine macrophages and DCs stimulated by LPS, the signaling of mammalian target of rapamycin complex 1 (mTORC1), whose activation depends on available nutrients including glucose, is sustained, and HIF-1α is upregulated, thus increasing glycolysis and triggering iNOS expression with consequences on OXPHOS as aforementioned [
16,
39,
48]. Moon et al. show that Raptor/mTORC1 complex is involved in the regulation of HK1 expression and glycolysis that regulates NLRP3 inflammasome activation [
39]. This glucose-sensitive signal transduction circuit coordinates DC metabolism and function to limit DC-stimulated T-cell responses [
48]. In glucose-deprived cells, this mTORC1/HIF-1α/iNOS pathway is impaired, thus impacting both DC metabolism and immunological functions [
35]. In these cells, NF-κB- and ERK-dependent transcriptional events, which are induced upon TLR engagement, are also required to trigger HIF-1α accumulation [
63,
64]. mTORC2 also enhances glycolytic metabolism by activating AKT and promoting MYC transcription activation [
65]. In addition, PKM2 can associate and regulate HIF-1α activity with consequences on IL-1β induction in LPS-activated macrophages [
30]. We have shown that quite differently, the increased expression of HIF-1α observed in human MoDCs stimulated by TLR4 depends on p38-MAPK activation [
7]. HIF-1α then increases HK2 levels in both human MoDCs and mouse BMDCs, resulting in higher HK activity and glycolytic flux [
7,
9]. In human MoDCs, cytokine secretion triggered by TLR4 stimulation depends on this p38-MAPK/HIF-1α/HK2 pathway, while other pathways are controlling the induction of maturation markers such as MHC-class II, CD40, and CD86 [
7]. Although TLR1/2 or TLR2/6 stimulation also results in a glycolytic burst in human MoDCs, the molecular mechanisms involved are not dependent on p38-MAPK activation of HIF-1α [
7]. Beyond its impact on glycolysis, this upregulation of HK2 has consequences on apoptosis and autophagy due to the nonenzymatic functions of this protein. Overexpression and mitochondrial association of HK2 confer protection to apoptotic or necrotic stimuli in different cell types by several mechanisms [
66]. Moreover, in response to glucose starvation, HK2 binds and inhibits mTORC1, thus facilitating autophagy [
66]. Although established in non-immune cells, this mechanism could contribute to protecting DCs or macrophages from cellular damage, providing energy by recycling intracellular components, but also contribute to internalized pathogen processing, antigen presentation and immune activation.