The aryl-hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates numerous cellular responses. Originally investigated in toxicology because of its ability to bind environmental contaminants, AhR has attracted enormous attention in the field of immunology in the last 20 years. In addition, the discovery of endogenous and plant-derived ligands points to AhR also having a crucial role in normal cell physiology. Thus, AhR is emerging as a promiscuous receptor that can mediate either toxic or physiologic effects upon sensing multiple exogenous and endogenous molecules. Within this scenario, several factors appear to contribute to the outcome of gene transcriptional regulation by AhR, including the nature of the ligand as such and its further metabolism by AhR-induced enzymes, the local tissue microenvironment, and the presence of coregulators or specific transcription factors in the cell.
- Aryl-hydrocarbon Receptor
- Matabolites
- coactivators
- dendritic cells
- immune regulation
1. Introduction
The aryl-hydrocarbon receptor (AhR) is a ligand-activated transcription factor that can recognize a large variety of molecules of both endogenous and exogenous origin. Because of this property, AhR acts as a crucial sensor of the cellular presence of a myriad of compounds capable of crossing the cell membrane [1,2]. The list of AhR ligands is impressive and striking in its diversity. Due to this ligand’s promiscuity and consequent pleiotropic functions, AhR plays critical roles in several physiologic processes such as xenobiotic metabolism, immune responses, cell proliferation, differentiation, and apoptosis [3,4]. Moreover, AhR involvement has been reported in the development and maintenance of several pathological conditions [3].
The different outcomes of AhR activation may depend on the specific ligand, the environment (i.e., conditions which can lead to the concomitant activation of other cellular pathways), and the interaction with specific comodulators of gene transcription and/or other transcription factors as well [5]. Thanks to several studies [6,7,8,9,10,11], the role of AhR has particularly emerged in the cells of the immune system, in which the receptor acts to balance the effector and regulatory arms of responses and thus represents an attractive target for future strategies of therapeutic immune modulation.
2. AhR, a Promiscuous Ligand-Activated Transcription Factor Regulating Immune Responses
2.1. Canonical AhR Activation by Endogenous and Exogenous Ligands
AhR can be activated by a structurally diverse spectrum of synthetic and environmental chemicals, including dietary components, microbiota-derived factors, and endogenous tryptophan (Trp) metabolites. The prototypical AhR exogenous ligand is represented by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), an environmental contaminant whose signaling has been largely studied. Halogenated dioxins, polycyclic aromatic hydrocarbons, and halogenated biphenyls represent the best characterized, high affinity planar aromatic and hydrophobic ligands of AhR. For a long time, it was believed that only molecules with specific physiochemical characteristics, such as hydrophobic structures with planar shape, could interact with AhR [12]. More recent research has shown that this “dogma” is not entirely correct. Indeed, a large number of structurally diverse natural, endogenous, and synthetic AhR ligands, having little similarity with prototypical AhR ligands, can activate the receptor [13]. Moreover, the discovery of these novel compounds able to interact with AhR, many of which have an endogenous source, has prompted more interest in the study of the major physiological roles of AhR. At present, the more deeply studied AhR endogenous ligands are those derived from Trp. The three major Trp derivatives that appear to contribute to the outcome of gene transcriptional regulation by AhR include kynurenines [14] in mouse lymphoid tissue dendritic cell (DCs), 6-formylindolo[3,2-b]carbazole (FICZ) in human skin keratinocytes [15], and microbiota-derived indole-3-aldehyde (IAld) in innate lymphoid cells of the gut in mice [16]. Although characterized by different physiochemical characteristics, these ligands are able to interact with AhR within the ligand binding domain, thus causing a conformational change translating into AhR transcriptional activity (Figure 1).
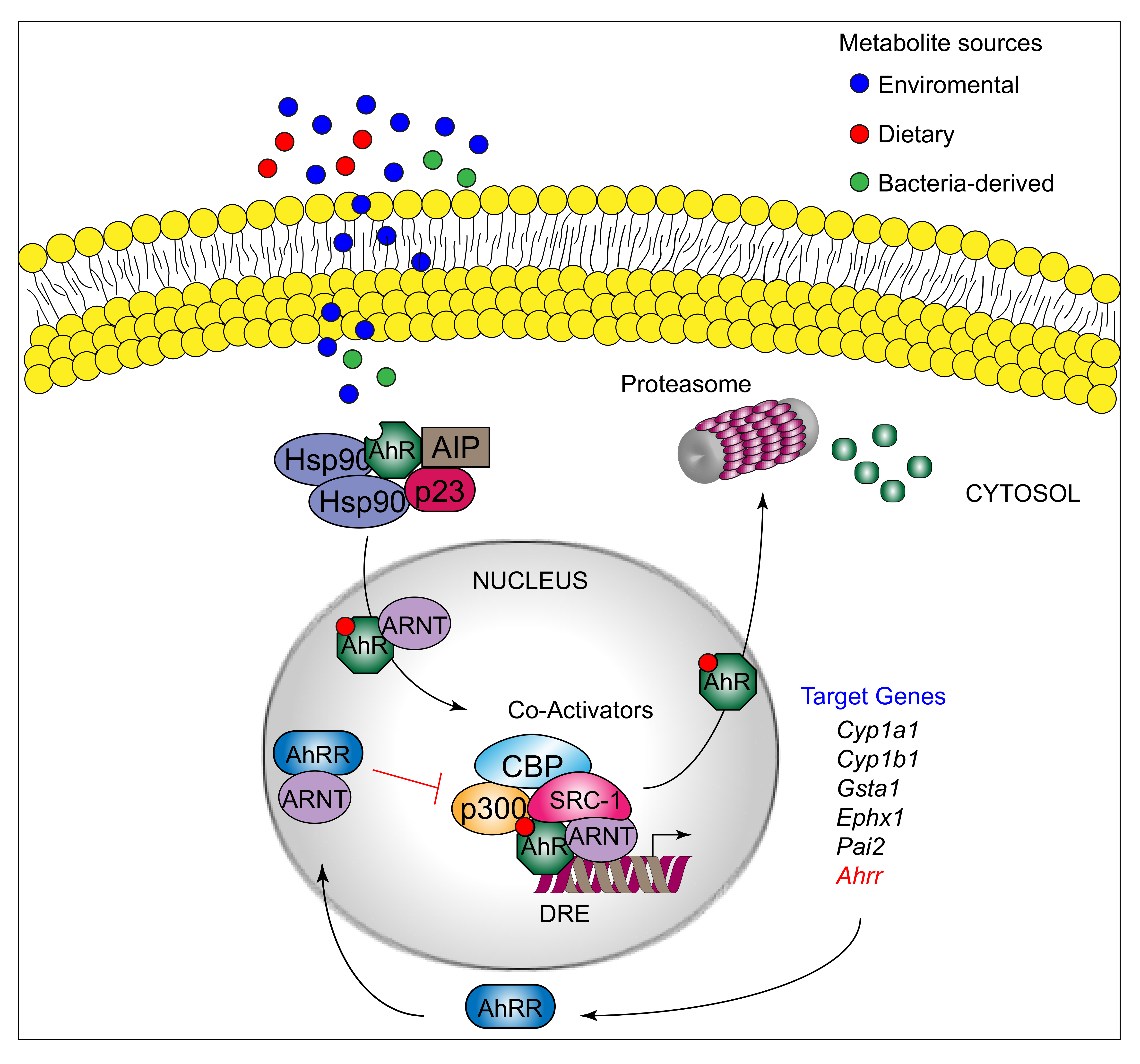
AhR belongs to the bHLH-PAS family of structurally related proteins, as it contains a basic Helix–Loop–Helix motif in the N-terminus and a PER/ARNT/SIM-homology domain in the C-terminal adjacent region. The PAS domain contains two imperfect repeats of 50 amino acids, namely, PAS A and PAS B, and is thought to serve as an interactive surface for dimer formation; in particular, PAS B represents the ligand-binding domain. The C-terminal half amino acidic sequence mediates transactivation activity through several specific sites, for instance a glutamine-rich box [17]. More specifically, the transactivation domain (TAD) facilitates the dynamic interplay between AhR and coactivators or coregulators [18,19]. In its inactive form, AhR is sequestered in the cytosol as part of a large complex containing several proteins, including the molecular chaperone HSP90 [20], the cochaperone p23 [21], the protooncogene SRC (c-SRC) tyrosine protein kinase [22], and the hepatitis B virus X-activating protein 2 (XAP2) [23] (also known as AhR-interacting protein or ARA9) [24,25]. The interaction of mammalian AhR with selected ligands induces the detachment of the receptor from this complex, leading to AhR nuclear translocation and promotion of AhR transcriptional activity [26] Specifically, the interaction of AhR with an agonist will induce conformational changes that trigger the receptor to dissociate from the chaperon complex, an event that drives AhR binding to the AhR nuclear translocator protein (ARNT) in the nucleus. However, evidence demonstrating that AhR may not necessarily dissociate from HSP90 to translocate to the nucleus has also been provided in human cells [27] In the nucleus, the AhR/ARNT complex interacts with multiple consensus sequences in the promoter of several genes, designated as xenobiotic responsive elements (XREs; also AhRREs and dioxin responsive elements, DREs). These events require ATP-dependent chromatin remodeling factors (e.g., BRG-1) and the recruitment of coactivators, such as the CBP/p300 histone-acetyltransferases by the ARNT moiety, and RIP140 and SRC-1 by the AhR fraction, which transmit the transactivation activities of the heterodimer [28]. In addition, other DNA elements cis-acting with XREs allow the AhR/ARNT complex to operate in synergy with other transcription factors. This control of transcriptional activity is known as the “canonical” genomic pathway of AhR signaling [29]. Different AhR ligands in specific cell compartments may trigger the formation of specific AhR protein complexes, which differ in their composition, leading to differential gene expression [30]. In addition to the genomic functions, several studies have shown that AhR regulates cellular responses through ARNT-independent, nongenomic mechanisms that involve Ca2+, c-SRC, cytosolic phospholipase A2 (cPLA2), and COX-2. In particular, after human and mouse AhR activation, c-SRC is released from the AhR/HSP90/c-SRC complex and thus acquires the ability to phosphorylate multiple cellular targets [31,32]. Moreover, it has been reported that AhR can act as a ligand-dependent E3 ubiquitin ligase, thus leading to the degradation of AhR-interacting proteins through the proteasome [33].
2.2. AhR Expression and Functions in Immune Cells: A Territory for Exploring the Intricacies of AhR Interaction with Coregulators?
AhR was originally identified in mouse cells and next identified also in human tissues, where the highest expression was found to be similar to mouse cells in the lung, thymus, kidney, and liver [34]. The elevated expression levels of AhR in barrier organs such as the skin, gut, and lung and the ability of AhR to bind many exogenous and endogenous compounds have recently attracted the attention of immunologists [7,10,35]. Although AhR-deficient mice do not exhibit a specific phenotype in steady state conditions, studies over the last 20 years have revealed that exposure of AhR deficient mice to inflammatory stimuli determines substantial immune defects in the body’s main barrier sites, such as skin [36,37], the lungs [38,39], and the gastrointestinal tract [40].
This evidence highlights the role of AhR as a gatekeeper of both physical and immunological barriers. The crucial importance of the AhR pathway in the regulation of the intestinal immune system has been demonstrated by genome-wide association studies that have identified the AHR gene as a susceptibility locus in inflammatory bowel diseases [41]. In fact, ablating AhR in CD11c+ cells (i.e., mostly represented by DCs) perturbs intestinal epithelium development and intestinal immunity [42]. Moreover, AhR plays an essential role in murine RORγt(+) ILC3, (type 3 innate lymphoid cells) maintenance and function, cells which are essential for gut immunity presumably through the production of interleukin-22 (IL-22) [40]. A recent study unveiled that AhR expression in epithelial intestinal cells is responsible for the metabolic clearance and detoxification of AhR ligands via induction of P4501 (CYP1) enzymes and that this negative feedback mechanism protects from excessive AhR activation [43]. Therefore, an AhR-mediated circuitry appears to be mandatory for the development and immunologic homeostasis of the gut.
Earlier studies demonstrated the crucial role of AhR in adaptive immune responses for both T(reg) and T(H)17 cell differentiation in a ligand-specific fashion, pointing to a unique target for therapeutic immunomodulation [44]. Intriguingly, it is now emerging that the AhR expression profile can change during the course of differentiation and activation in many immune cells. In this regard, a typical example is constituted by T cell subsets. In fact, AhR is more expressed in mouse T helper type 17 (Th17) cells compared to non-polarized activated Th0 cells as well as to Th1, Th2, and regulatory T (Treg) cells [45]. Moreover, numerous studies have reported the expression of AhR in several other immune cell types including B cells, mucosal cells, and antigen-presenting cells (APCs) [10].
With respect to AhR ligands, the controversial effects of AhR agonists have been observed in the control of T cell function, especially in autoimmune diseases. For example, TCDD treatment of CD4+ T cells results in an AhR-dependent induction of Foxp3+ Treg cells in vitro, and TCDD administration in mice with experimental autoimmune encephalomyelitis (EAE; a model of multiple sclerosis) results in Treg cell expansion and suppression of the disease [44]. In contrast, Veldhoen et al. demonstrated that FICZ, locally administered during EAE, enhances the differentiation of Th17 cells [45], while in other studies the systemic administration of FICZ significantly reduces the signs of EAE [46]. These data strongly highlight the role of tissue microenvironment in conditioning the outcome of AhR activation in specific immune cells.
Several cells have been reported to express AhR, mostly correlating with immunoregulatory phenotypes. Specifically, a crucial role of AhR has been reported for human type 1 regulatory T cells (Tr1), in which AhR, in concert with c-Maf, controls IL-21 and IL-10 production [47]. Different levels of AhR are also expressed in different subsets of mouse and human B lymphocytes [48,49,50]. In 1990, Kerkvliet et al. reported that AhR activation by environmental toxins can be responsible for the suppression of humoral immune responses [51]. More recent studies demonstrated that B-cell receptor activation leads to AhR induction in B cells, and that AhR-deficient B cells proliferate less well than their wild-type counterparts [52]. Microarray analysis of myeloid cells indicated AhR expression also in both macrophages and DCs [35], which was also confirmed by data on AhR protein expression both in mouse and human counterparts [14,53,54,55]. The levels of AhR expression increase upon LPS and other Toll-like receptor (TLR) ligand engagement in macrophages and DCs and B cells [32,56,57,58]. Specifically, recent studies demonstrated that AhR regulates the differentiation and functions of CD19+CD21hiCD24hi regulatory B (Breg) cells, producing IL-10 and thus limiting the differentiation of inflammatory B cells [57]. Interestingly, AhR-deficient APCs also show a defect in IL-10 production [59].
The role of AhR in regulating DC functions has been extensively studied. At present, it is known that AhR activation alters DC differentiation and innate functions. Bone marrow-derived DCs (BMDCs) from C57BL/6 mice, generated in the presence of TCDD, decrease CD11c expression and increase MHC class II and CD86 expressions [60]. Similar effects were shown for other AhR ligands, such as indole-3-carbinol (I3C) and indirubin-3′-oxime (IO). Both I3C and IO decrease the expression of CD11c, CD40, and CD54, while increasing the expression of MHC class II and CD80. Moreover, following lipopolysaccharide (LPS) activation, I3C and IO suppress the production of pro-inflammatory mediators, including TNF-α, IL-6, IL-12, and nitric oxide but increase IL-10 levels. Additionally, immunoregulatory genes, such as those coding for aldehyde dehydrogenase (ALDH1A), indoleamine 2,3-dioxygenase 1 (IDO1), and TGF-β are up-regulated after treatment with I3C or IO [61]. Moreover, it was also demonstrated that activation of AhR in DCs is able to induce SOCS2 expression, thus limiting the production of pro-inflammatory IL-6 and IL-12 [62]. Concomitantly, DC activation by the AhR ligands TCDD and the tryptophan metabolite kynurenine promotes the expression of IDO1 and IDO2 immunoregulatory enzymes [14] which, in turn, by catalyzing further production of kynurenine, sustain the induction of TGF-β and lead to the acquisition of tolerogenic properties by the DCs [32,59].
Overall, although AhR is expressed in several immune cell types, the outcome of AhR activation by specific ligands may be different. This difference may be ascribed to the presence of AhR partners other than prototypical ARNT in different cell types.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22020757