Cardiac connexin-43 (Cx43) creates gap junction channels (GJCs) at intercellular contacts and hemi-channels (HCs) at the peri-junctional plasma membrane and sarcolemmal caveolae/rafts compartments.
- cardiac arrhythmias
- gap junction channels
- Cx43 hemichannels
- Panx1 channels
1. Introduction
Cx43 is the most expressed cardiac protein in the heart out of all the 20 rodents and 21 human connexin isoforms. As a typical integral membrane protein, Cx43 is synthesized in the rough endoplasmic reticulum and translocated into the Golgi compartment (Figure 1a) for folding and oligomerization into connexons. These are termed hemichannels (HCs) and they then follow the secretory pathway to reach their final destination at the plasma membrane[1][2][3][4][5][6][7][8][9][10][11][12] [27,28,29,30,31,32,33].
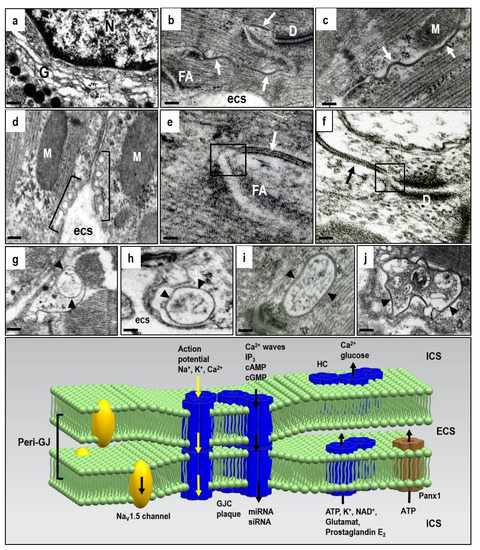
Figure 1. Electron microscopic images of the adult rat heart cardiomyocytes showing structures hosting Cx43. (a)—Golgi compartment in which Cx43 is oligomerized to HCs; (b–d)—post-Golgi HCs underwent targeted delivery to the particular membrane-subdomains, i.e., caveolae/rafts and gap junctions, close apposition of neighboring cardiomyocytes, which consist of paired HCs forming gap junction channels (GJCs). Gap junction plaques (arrows) are prevalent at the intercalated disc (b) and scarce at the lateral sides of the cardiomyocytes (c), while caveolae/rafts (brackets) are abundant at the lateral membranes (d). Peri-junctional plasma membranes (boxes in (e,f)) contain undocked HCs and Panx1 channels. The internalization of gap junctions into annular profiles (arrowheads in g–j) precedes degradation of HCs via proteasomal and lysosomal pathways. Abbreviations: G—Golgi compartment, N—nucleus, D—adhesive junction, desmosome, FA—fascia adherent junction, M—mitochondria, esc—extracellular space, scale bar—0.1 micrometer. The diagram demonstrates the putative topology of GJCs at the gap junction plaque, HCs and Panx1 and also the NaV1.5 channels at the peri-junctional membranes together with transmitted ions and molecules; ICS—intracellular space, ECS—extracellular space. Tribulova, unpublished representative electron micrographs.
The mechanism of how newly translated oligomerized proteins arrive at their proper location is still being explored[1][10][13] [27,31,34].
Docking two HCs from neighboring cardiomyocytes into GJCs and assembly into gap junction plaques is quite prevalent at the intercalated discs, but rare at the lateral plasma membrane of cardiomyocytes in the healthy heart (Figure 1b,c). Connexin is characterized by four transmembrane segments with α-helical conformation and N- and C-terminal tails projecting into the cytoplasm. Two extracellular loops (E1 and E2), containing about 31 and 34 amino acids, respectively, are highly conserved and covalently connected by three invariant disulphide bonds. The extracellular domains E1 and E2 of the Cx43 have a critical function in the formation of the complete GJCs. Early phosphorylation and the presence of adhesion molecules appears essential prerequisites for HCs to GJCs assembly[1][2][3] [27,28,29].
Some HCs in peri-junctional membranes (peri-nexus) do not undergo docking, and exert specific non-canonical functions beyond GJCs[4][13][14] [25,34,35]. However, their mutual proximity implies integrated HCs and GJCs interaction and cooperation (Figure 1e,f). In addition, the peri-junctional membranes contain clusters of Nav 1.5 channels that influence cardiac conduction[15] [36], and these are combined with the large non-selective conductance channel constituted by pannexin1 (Panx1) [1][2][3][16][17][22,23,24,37,38]. There is also evidence for an ephaptic contribution to cardiac conduction which influences synchronization and timing of action potential firing [18][19][20][39,40,41]. The ephaptic conduction of electrical impulses occurs through transients in extracellular ions at the intercalated disc, and this prompts the activation of adjacent myocytes. This mechanism, however, appears to be localized in the peri-junctional region, and it implicates HCs and Panx1 channel activation.
Pannexins form channels at the plasma membrane surface that establish a pathway for communication between the cytosol of individual cells and their extracellular environment. Pannexins do not form intercellular GJCs, but only membrane channels. The pannexin family consists of 3 members, namely Panx1, Panx2, and Panx3, whereby Panx1 is the most expressed in the heart[21][22][23] [42,43,44]. Panx1 was found to bind directly to actin, and this suggest that microfilaments act to stabilize Panx1 cell surface distribution or even participate in Panx1 delivery or internalization[17] [38]. The current destination of HCs can also be determined by specific lipid rafts or membrane micro-domains associated with caveolae at the sarcolemma (Figure 1d). These are most important in compartmentalizing a variety of signaling activities[24] [45]. Both HC and GJC are recycled via a post-endocytic event and this is consistent with rapid myocardial Cx43 turnover and very short half-life of 1–2 h [25][26][46,47]. GJCs from the middle part of the gap junction plaque internalize to form a circular profile as “annular gap junctions” (Figure 1g). These undergo degradation by proteasomes and lysosomes[5][8][9][10][25] [27,29,30,31,46]. However, the considerably rapid HCs/GJCs turnover is most important because it illustrates the dynamic nature of intercellular communication and Cx43 mediated signaling [4][5][27][14][16][27][25,27,35,37,48]. In contrast to the Cx43 protein, the Panx1 channels are composed of long-lived pannexin proteins which suggests a slower life cycle where internalized pannexins are degraded by lysosomes[17] [38].
In addition to evidence that GJCs and HCs promote opposite responses in cellular signaling events, molecular studies indicate differences in GJCs’ and HCs’ dependence on intramolecular loop/tail interactions for their activity [27][28][29][30][35,49,50,51]. Monoclonal antibodies to the extracellular loop E2 enable HCs visualization of cell membranes without GJCs visualization because E2 epitope is not free in dodecameric GJCs[31][32] [52,53]. These antibodies could aid HCs topology study through the changes in their myocardial distribution in response to acute and chronic heart injury associated with arrhythmias.
As illustrated in the diagram in Figure 1, GJCs, HCs and the Panx1 channels create a unique and dynamic signaling network in the heart and vascular system to ensure tissue homeostasis and proper function. This network coordinates myocardial contraction. It is also the most important in heart adaptation, and especially for the response to stressful or pathophysiological conditions by propagating both cell survival and apoptosis signals [33][34][35][16][17][25][30][36][20,22,25,37,38,46,51,54]. It is a fundamental and permanent task to control this network when trying to prevent or treat heart and life-threatening arrhythmias. Evidence suggests that pharmacological HCs and Panx1 channel blocking prevents their undesirable action following stress or injury, and also that the protective GJCs function could provide new treatment opportunities.
2. Implication of HCs and Panx1 Channels in Disturbances of Ionic and Redox Homeostasis as Well as Pro-Inflammatory and Pro-Fibrotic Signaling
Compared to GJCs, the HCs and Panx1 channels operate out of cardiomyocyte membrane gap junctions. Therefore, dominant myocardial HCs and Panx1 channel topology appears restricted to the lipid rafts/caveolae on lateral cardiomyocyte plasma membrane (Figure 1d). In addition, they also act at the peri-nexus [34][22], i.e., at the peri-gap junctional membrane (Figure 1e,f). HCs also perform the “non-canonical” functions of Cx43, beyond those of the GJCs and independent of gap-junction formation [16][37][38][25,55,56].
Findings from various non-cardiac tissues that might be relevant for cardiomyocytes indicate that HCs channels remain closed in physiological resting conditions, but change to open conformation as the extracellular Ca2+ concentration decreases or intracellular Ca2+ concentration increases, which open Panx1 channels as well[39][40][41][42][43] [57,58,59,60,61] However, HCs and Panx1 channels do not have the exact same regulation. HCs tend to be activated by strong depolarization, while Panx1 channels activity may be induced at the resting membrane potential[44] [62]. The pore diameter increases from 1.8 to 2.5 nm and this enables transmission of molecules up to 1 kD. HCs opening enables typical single-channel conductance of approximately 220 pS[45] [63] and Panx1 channels have a large unitary conductance of around 500 pS. In addition, hydrophobic extracellular domains are also crucial in regulating Ca2+-dependent conformational changes[33] [20].
The HCs and Panx1 channels can open following different extracellular stimuli. This is established in various non-cardiac tissues[39][40][41][42][43][44][45][46][47] [57,58,59,64,65], but highly relevant to the heart. The HCs and Panx1 channels can therefore be activated by membrane depolarization, metabolic inhibition, and stresses including shear mechanical membrane forces and ionic and ischemic stress[3][17][48][49][24,38,57,62,66,67]. The HCs, and most likely also the Panx1 channels, are influenced by intra- and extra-cellular pH and phosphorylation and redox status[34][3][16][40][48][50][51][52] [22,24,37,58,66,68,69,70]. All these events during heart disease development can activate both HCs and Panx1 channels and promote arrhythmias through bi-directional ion passage and small metabolic or signaling molecules below 1–2 kDa. Thus, HCs and/or Panx1 channels can mediate Na+, K+, Ca2+, cAMP, ATP, NAD+ influx as well as the transmission of glutamate, glutathione, prostaglandin-E2 and epoxyeicosatrienoic acid [3][14][53][24,35,71]. Panx-1 channels are known mostly as ATP release channels[42] [60]. Intracellular localized Panx1 has also been proposed as a regulator of sarcoplasmic reticulum-based Ca2+ homeostasis[54] [72].
HCs and Panx1 channel opening can profoundly affect intracellular ionic homeostasis, contribute to Na+ and Ca2+ loading and K+ loss, alter redox status, and facilitate pro-inflammatory and pro-fibrotic conditions [36][39][40][55][56][54,57,58,62,73]. These events impair GJCs function and produce myocardial electrical instability, thereby promoting malignant arrhythmias [57][11][48][58][59][4,32,66,74,75]. It was also previously noted that the activation of peri-junctional Panx1 channels [16][37] and Nav 1.5 channels can influence cardiac conduction [36,39]. Abnormal Nav1.5 channel function along with autoantibodies against Cx43 and activation of Panx1 channels may underlie heritable arrhythmic syndromes[60][61][62] [76,77,78].
HCs and Panx1 channel mediated ATP release is fundamental in the purinergic signaling which is so important in promoting inflammation in vascular function/dysfunction[44][63][48][64][65][66] [62,65,66,79,80,81]. The inflammation is known to impair GJCs function and significantly contribute to arrhythmogenesis [74]. The Panx1 channels present at internal membranes may be implicated in K+ influx to mitochondria and intracellular Ca2+ leak/release from the endoplasmic reticulum[27][53][67] [35,71,82]. Moreover, ATP binding to the P2Y receptors increases inositol 1,4,5-triphosphate which then releases Ca2+ from the ER and enhances ATP signaling to neighboring cells[1] [24]. Disturbances in intracellular ionic homeostasis and Ca2+ overload promote triggered “post-depolarization” and GJCs uncoupling which render the heart prone to malignant arrhythmia development; and especially VF [68][69][70][71][72][83,84,85,86,87]. This triggered Cx activity and GJCs disordered expression and dysfunction are also implicated in AF development[70][73] [85,88].
Cellular stress, including myocardial ischemia, enhances cardiomyocyte ATP release through the Panx1 channels that facilitate early fibroblast activation[42] [66]. This suggests an early paracrine event leading to pro-fibrotic responses which could be involved in arrhythmogenic substrate development. In contrast, HCs inhibition by TAT-Gap19 alleviated tissue fibrosis and introduced the possibility of preventing arrhythmias[74] [89]. Fibrosis precedes inflammation, and cardiac fibroblasts contribute to the inflammatory milieu through increased secretion of pro-inflammatory cytokines and chemokines released by the HCs and Panx1 channels[75][76] [90,91]. Here, the Panx1 channels are interesting because of their implication in a variety of cellular responses[3][17] [24,38], including apoptosis [77][92], inflammation [78][93], and innate immune processes [79][80][94,95]. Inflammation is also a prominent feature of arrhythmogenic cardiomyopathy[81] [96]. Therefore, targeting inflammatory pathways could be an effective new mechanism-based therapy for familial non-ischemic heart muscle disease which causes sudden death in the young, and especially in athletes.
Figure 2 illustrates that functional HCs and Panx1 channels provide paracrine and autocrine communication pathways for ion and small molecule passage across the plasma membrane[1][16][17][48][64] [22,37,38,66,97]. HCs and Panx1 channels may regulate many cellular processes but little information on their mechanisms is available. Extended information on HCs and Panx1 channel function from in vitro sources indicates their implication in diverse physiological and pathological responses relevant to the cardiovascular system[33][1] [20,22]. However, the cardiac muscle remains ‘terra incognita’ and this provides challenges. This is especially important, in conjunction with rafts/caveolae related signal-transducing molecules, in the development of heart disease and life-threatening arrhythmias. The compartmentalized signal transduction appears to be an attractive area of research.
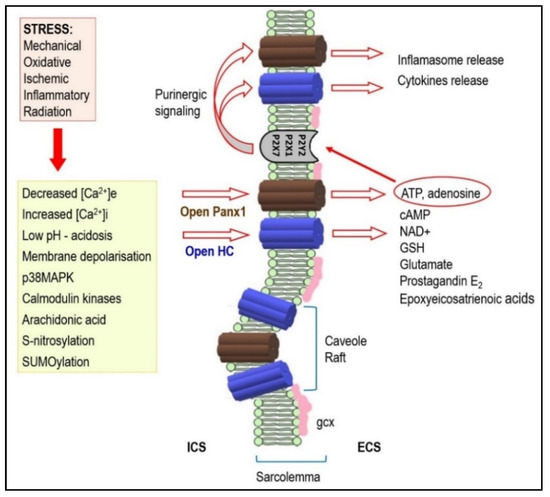
Figure 2. Under stressful conditions, several intracellular signals regulate hemi-channels (HCs) opening. Activated HCs enables release of signaling molecules to the extracellular environment. Therefore, it acts as a paracrine and autocrine communication pathway. HCs mediated ATP release is important in the purinergic signaling which is so important in cardiac pathophysiology. It may also be implicated in arrhythmogenesis (see text for details). Abbreviations: ICS—intracellular space, ECS—extracellular space, gcx—glycocalyx.
It is important that the caveolae and lipid rafts of cholesterol and sphingomyelin enriched membrane micro-domains are considered in HCs and Panx1 channels. These are all involved in the regulation of cell signaling pathways [40][41][82][83][58,59,98,99] and in GJCs homeostasis[84][85][86][87][88] [100,101,102,103,104]. Caveolin-3 is the major caveolin isoform in cardiomyocytes, and this has been implicated in 17β-estradiol-elicited, rapid signaling to regulate Cx43 phosphorylation during ischemia[85] [101]. Moreover, the caveolae/lipid rafts have been involved in reactive oxygen species production, redox signaling and K+, KATP, Na+, and Ca2+ ion channel functions [89][90][91][105,106,107].
Cardiomyocytes and endothelial capillary cells are rich in caveolae which invaginate the plasma membrane, and this suggests the implication of these compartments in extensive HCs and Panx1 channel-mediated paracrine and autocrine signaling. The interaction of these channels and caveolins implies compartmentalized signaling, and this could be most important in the pathophysiological development of heart dysfunction and arrhythmias [24][87][88][92][93][94][45,103,104,108,109,110]. Investigation of this issue presents future challenges.
3.Impact ofHCs and Pannex1 Channels Activity onDevelopment of Cardiac Arrhythmias
There is still a lack of information on HCs and Panx1 channel roles in cardiac muscle. Nevertheless, it has been shown that both HCs and Panx1 channels are the potential route for Na+ inflsux and K+ efflux during ischemia in isolated ventricular cardiomyocytes [39,40,62,128]. This can induce electrical disturbances [95][20][14,41]. The opening of HCs and Panx1 channels during ischemia has also contributed to re-perfusion injury following brief cardiomyocyte ischemia[48][76][96][97][98] [66,91,129–131]. The activation of HCs and Pannex1 channels during ischemia/hypoxia, combined with other metabolic inhibition and post-ischemic reperfusion mechanisms may compromise cardiomyocyte ability tomaintain ionic homeostasis. This is an essential step in promoting both arrhythmias and apoptosis[72][76][98][99][100][101] [87,91,131–134]. Metabolic inhibition followed by pro-arrhythmogenic [Ca2+]i and [Na+]i overload in isolated cardiomyocytes was significantly reduced by halothane which decreases HCsconductance [102][135]. Importantly, HCs contribute to cytoplasmic Ca2+ oscillations by providinga bimodal Ca2+ dependent Ca2+ entry pathway[103] [136]. Moreover, ischemia and/or hypoxia are associated with increased production of NO and S-nitrosylation [104][105][137,138] which is important in regulating HCs and Panx1 channel permeability[106][107] [139,140]. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential[108] [141]. The NADPH oxidase inhibitor apocynin prevented HCs activity by reducing nitroso-redox stress [109][142]. All these factors strongly suggest that HCs are implicated in arrhythmogenesis.
The release of ATP via HCs or Panx1 channels in pathological conditions could induce post-depolarization triggered activity. This would generate ventricular tachycardia or VF[110][111] [145,146]. Moreover, purinergic receptor activation by ATP induces ventricular tachycardia through membrane depolarization and Ca2+ homeostatic disorders[112] [147]. Resultant sporadic Panx1 channel opening then triggered action potentials and promoted arrhythmogenic activity[2] [23].
In addition, extracellular ATP induced shortened action potential duration in tissue preparations of atrial and ventricular myocardium and in the myocardial sleeves of pulmonary veins[113] [148]. These events are known to precede AF[70][114] [85,117]. The implication of ATP or adenosine in AF development, and its post-ablation recurrence, have been
reported[115][116][117][118][119] [149–153]. This AF was associated with a strong increase in atrial adenosine[120] [154].
In addition, non-myocytes in scar tissue can be electrically coupled to cardiomyocytes through GJCs, HCs, and Panx1 channels and contribute to electrotonic conduction across scar tissue[121] [122][158,159]. Cx43 is increased in activated fibroblasts during fibrogenesis[76][123][124] [91,160,161] and it enhances the possibility of electrically coupled cardiomyocytes and fibroblasts [123][124][125][160–162]. This hetero-cellular electrotonic coupling may facilitate conduction disturbances and arrhythmias [126][163]. Fibroblasts and myofibroblasts can alter cardiomyocyte excitation-contraction coupling through paracrine mechanisms[127] [164], and this suggests the paracrine modulation of myocardial contractility and synchronized contraction. Fibrotic tissue, however, is an inefficient electrical signal conductor and cannot rhythmically contract, thereby jeopardizing critical myocardial functions [91]. This combination provides heterogeneous cardiac tissue, and this is a well-known predictor of arrhythmia risk. Although HCs and Panx1 channels present an attractive therapeutic target, their inhibition requires the development of more specific inhibitory agents.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010260
References
- Meens, M.J.; Kwak, B.R.; Duffy, H.S. Role of connexins and pannexins in cardiovascular physiology. Cell. Mol. Life Sci. 2015, 72, 2779–2792, doi:10.1007/s00018-015-1959-2.
- Kienitz, M.C.; Bender, K.; Dermietzel, R.; Pott, L.; Zoidl, G. Pannexin 1 constitutes the large conductance cation channel of cardiac myocytes. J. Biol. Chem. 2011, 286, 290–298, doi:10.1074/jbc.M110.163477.
- Penuela, S.; Gehi, R.; Laird, D.W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta Biomembr. 2013, 1828, 15–22, doi:10.1016/j.bbamem.2012.01.017.
- Rusiecka, O.M.; Montgomery, J.; Morel, S.; Batista-Almeida, D.; Van Campenhout, R.; Vinken, M.; Girao, H.; Kwak, B.R. Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules 2020, 10, 1225, doi:10.3390/biom10091225.
- Mugisho, O.O.; Rupenthal, I.D.; Paquet-Durand, F.; Acosta, M.L.; Green, C.R. Targeting connexin hemichannels to control the inflammasome: The correlation between connexin43 and NLRP3 expression in chronic eye disease. Expert Opin. Ther. Targets 2019, 23, 855–863, doi:10.1080/14728222.2019.1673368.
- Evans, W.H. Cell communication across gap junctions: A historical perspective and current developments. Biochem. Soc. Trans. 2015, 43, 450–459, doi:10.1042/BST20150056.
- Salameh, A. Life cycle of connexins: Regulation of connexin synthesis and degradation. Adv. Cardiol. 2006, 42, 57–70, doi:10.1159/000092562.
- Su, V.; Lau, A.F. Connexins: Mechanisms regulating protein levels and intercellular communication. FEBS Lett. 2014, 588, 1212–1220, doi:10.1016/j.febslet.2014.01.013.
- Solan, J.L.; Lampe, P.D. Spatio-temporal regulation of connexin43 phosphorylation and gap junction dynamics. Biochim. Biophys. Acta Biomembr. 2018, 1860, 83–90, doi:10.1016/j.bbamem.2017.04.008.
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta Biomembr. 2018, 1860, 40–47, doi:10.1016/j.bbamem.2017.05.018.
- Smyth, J.W.; Hong, T.T.; Gao, D.; Vogan, J.M.; Jensen, B.C.; Fong, T.S.; Simpson, P.C.; Stainier, D.Y.R.; Chi, N.C.; Shaw, R.M. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. J. Clin. Investig. 2010, 120, 266–279, doi:10.1172/JCI39740.
- Sarma, J. Das; Kaplan, B.; Willemsen, D.; Koval, M. Identification of rab20 as a potential regulator of connexin43 trafficking. Cell Commun. Adhes. 2008, 15, 65–74, doi:10.1080/15419060802014305.
- Sáez, J.C.; Leybaert, L. Hunting for connexin hemichannels. FEBS Lett. 2014, 588, 1205–1211, doi:10.1016/j.febslet.2014.03.004.
- Iyyathurai, J.; D’Hondt, C.; Wang, N.; De Bock, M.; Himpens, B.; Retamal, M.A.; Stehberg, J.; Leybaert, L.; Bultynck, G. Peptides and peptide-derived molecules targeting the intracellular domains of Cx43: Gap junctions versus hemichannels. Neuropharmacology 2013, 75, 491–505, doi:10.1016/j.neuropharm.2013.04.050.
- Hichri, E.; Abriel, H.; Kucera, J.P. Distribution of cardiac sodium channels in clusters potentiates ephaptic interactions in the intercalated disc. J. Physiol. 2018, 596, 563–589, doi:10.1113/JP275351.
- Scemes, E.; Spray, D.C.; Meda, P. Connexins, pannexins, innexins: Novel roles of “hemi-channels.” Pflugers Arch. Eur. J. Physiol. 2009, 457, 1207–1226, doi:10.1007/s00424-008-0591-5.
- Penuela, S.; Laird, D.W. The cellular life of pannexins. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 621–632, doi:10.1002/wmts.63.
- Rhett, J.M.; Ongstad, E.L.; Jourdan, J.; Gourdie, R.G. Cx43 associates with Nav1.5 in the cardiomyocyte perinexus. J. Membr. Biol. 2012, 245, 411–422, doi:10.1007/s00232-012-9465-z.
- Rhett, J.M.; Veeraraghavan, R.; Poelzing, S.; Gourdie, R.G. The perinexus: Sign-post on the path to a new model of cardiac conduction? Trends Cardiovasc. Med. 2013, 23, 222–228, doi:10.1016/j.tcm.2012.12.005.
- Veeraraghavan, R.; Lin, J.; Hoeker, G.S.; Keener, J.P.; Gourdie, R.G.; Poelzing, S. Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: An experimental and modeling study. Pflugers Arch. Eur. J. Physiol. 2015, 467, 2093–2105, doi:10.1007/s00424-014-1675-z.
- Penuela, S.; Harland, L.; Simek, J.; Laird, D.W. Pannexin channels and their links to human disease. Biochem. J. 2014, 461, 371–381, doi:10.1042/BJ20140447.
- Deng, Z.; He, Z.; Maksaev, G.; Bitter, R.M.; Rau, M.; Fitzpatrick, J.A.J.; Yuan, P. Cryo-EM structures of the ATP release channel pannexin 1. Nat. Struct. Mol. Biol. 2020, 27, 373–381, doi:10.1038/s41594-020-0401-0.
- Michalski, K.; Syrjanen, J.L.; Henze, E.; Kumpf, J.; Furukawa, H.; Kawate, T. The Cryo-EM structure of a pannexin 1 reveals unique motifs for ion selection and inhibition. Elife 2020, 9, 1–14, doi:10.7554/eLife.54670.
- Anderson, R.G.W. The caveolae membrane system. Annu. Rev. Biochem. 1998, 67, 199–225, doi:10.1146/annurev.biochem.67.1.199.
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635, doi:10.1161/01.RES.83.6.629.
- Martins-Marques, T.; Catarino, S.; Gonçalves, A.; Miranda-Silva, D.; Gonçalves, L.; Antunes, P.; Coutinho, G.; Leite Moreira, A.; Falcão Pires, I.; Girão, H. EHD1 Modulates Cx43 Gap Junction Remodeling Associated with Cardiac Diseases. Circ. Res. 2020, E97–E113, doi:10.1161/CIRCRESAHA.119.316502.
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15, doi:10.1016/j.abb.2012.03.008.
- Ponsaerts, R.; De Vuyst, E.; Retamal, M.; D’Hondt, C.; Vermeire, D.; Wang, N.; De Smedt, H.; Zimmermann, P.; Himpens, B.; Vereecke, J.; et al. Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J. 2010, 24, 4378–4395, doi:10.1096/fj.09-153007.
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64, doi:10.1016/j.bbamem.2017.05.008.
- García, I.E.; Sánchez, H.A.; Martínez, A.D.; Retamal, M.A. Redox-mediated regulation of connexin proteins; focus on nitric oxide. Biochim. Biophys. Acta Biomembr. 2018, 1860, 91–95, doi:10.1016/j.bbamem.2017.10.006.
- Baklaushev, V.P.; Gurina, O.I.; Yusubalieva, G.M.; Grinenko, N.F.; Cytrin, E.B.; Victorov, I.V.; Chekhonin, V.P. Immunofluorescent analysis of connexin-43 using monoclonal antibodies to its extracellular domain. Bull. Exp. Biol. Med. 2009, 148, 725–730, doi:10.1007/s10517-010-0802-x.
- Riquelme, M.A.; Kar, R.; Gu, S.; Jiang, J.X. Antibodies targeting extracellular domain of connexins for studies of hemichannels. Neuropharmacology 2013, 75, 525–532, doi:10.1016/j.neuropharm.2013.02.021.
- Rodríguez-Sinovas, A.; Sánchez, J.A.; Fernandez-Sanz, C.; Ruiz-Meana, M.; Garcia-Dorado, D. Connexin and pannexin as modulators of myocardial injury. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1962–1970, doi:10.1016/j.bbamem.2011.07.041.
- Meens, M.J.; Kwak, B.R.; Duffy, H.S. Role of connexins and pannexins in cardiovascular physiology. Cell. Mol. Life Sci. 2015, 72, 2779–2792, doi:10.1007/s00018-015-1959-2
- Rusiecka, O.M.; Montgomery, J.; Morel, S.; Batista-Almeida, D.; Van Campenhout, R.; Vinken, M.; Girao, H.; Kwak, B.R. Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules 2020, 10, 1225, doi:10.3390/biom10091225
- John, S.; Cesario, D.; Weiss, J.N. Gap junctional hemichannels in the heart. Acta Physiol. Scand. 2003, 179, 23–31, doi:10.1046/j.1365-201X.2003.01197.x.
- Spray, D.C.; Ye, Z.C.; Ransom, B.R. Functional connexin “hemichannels”: A critical appraisal. Glia 2006, 54, 758–773, doi:10.1002/glia.20429.
- Agullo-Pascual, E.; Delmar, M. The noncanonical functions of Cx43 in the heart. J. Membr. Biol. 2012, 245, 477–482, doi:10.1007/s00232-012-9466-y.
- Hervé, J.C. The communicating junctions, roles and dysfunctions. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1–3, doi:10.1016/j.bbamem.2012.10.012.
- Evans, W.H.; De Vuyst, E.; Leybaert, L. The gap junction cellular internet: Connexin hemichannels enter the signalling limelight. Biochem. J. 2006, 397, 1–14, doi:10.1042/BJ20060175.
- Schalper, K.A.; Palacios-Prado, N.; Orellana, J.A.; Sáez, J.C. Currently used methods for identification and characterization of hemichannels. Cell Commun. Adhes. 2008, 15, 207–218, doi:10.1080/15419060802014198.
- Dahl, G. ATP release through pannexon channels. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140191, doi:10.1098/rstb.2014.0191.
- Riquelme, M.A.; Cea, L.A.; Vega, J.L.; Boric, M.P.; Monyer, H.; Bennett, M.V.L.; Frank, M.; Willecke, K.; Sáez, J.C. The ATP required for potentiation of skeletal muscle contraction is released via pannexin hemichannels. Neuropharmacology 2013, 75, 594–603, doi:10.1016/j.neuropharm.2013.03.022.
- Taylor, K.A.; Wright, J.R.; Mahaut-Smith, M.P. Regulation of Pannexin-1 channel activity. Biochem. Soc. Trans. 2015, 43, 502–507, doi:10.1042/BST20150042.
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309, doi:10.1007/s00395-012-0309-x.
- Barria, I.; Guiza, J.; Cifuentes, F.; Zamorano, P.; Saez, J.C.; Gonzalez, J.; Vega, J.L. Trypanosoma cruzi infection induces pannexin-1 channel opening in cardiac myocytes. Am. J. Trop. Med. Hyg. 2018, 98, 105–112, doi:10.4269/ajtmh.17-0293.
- Sáez, J.C.; Green, C. Involvement of connexin hemichan
- Dolmatova, E.; Spagnol, G.; Boassa, D.; Baum, J.R.; Keith, K.; Ambrosi, C.; Kontaridis, M.I.; Sorgen, P.L.; Sosinsky, G.E.; Duffy, H.S. Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. Am. J. Physiol. Hear. Circ. Physiol. 2012, 303, 1208–1219, doi:10.1152/ajpheart.00251.2012.
- Kim, J.C.; Son, M.J.; Woo, S.H. Ca2+ Signaling triggered by shear-autocrine P2X receptor pathway in rat atrial myocytes. Cell. Physiol. Biochem. 2018, 50, 2296–2313, doi:10.1159/000495089.
- Ponsaerts, R.; Wang, N.; Himpens, B.; Leybaert, L.; Bultynck, G. The contractile system as a negative regulator of the connexin 43 hemichannel. Biol. Cell 2012, 104, 367–377, doi:10.1111/boc.201100079.
- Retamal, M.A.; Sáez, J.C. Hemichannels; from the molecule to the function. Front. Physiol. 2014, 5, 3389, doi:10.3389/fphys.2014.00411.
- Decrock, E.; Hoorelbeke, D.; Ramadan, R.; Delvaeye, T.; De Bock, M.; Wang, N.; Krysko, D.V.; Baatout, S.; Bultynck, G.; Aerts, A.; et al. Calcium, oxidative stress and connexin channels, a harmonious orchestra directing the response to radiotherapy treatment? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1099–1120, doi:10.1016/j.bbamcr.2017.02.007.
- D’hondt, C.; Ponsaerts, R.; De Smedt, H.; Vinken, M.; De Vuyst, E.; De Bock, M.; Wang, N.; Rogiers, V.; Leybaert, L.; Himpens, B.; et al. Pannexin channels in ATP release and beyond: An unexpected rendezvous at the endoplasmic reticulum. Cell. Signal. 2011, 23, 305–316, doi:10.1016/j.cellsig.2010.07.018.
- Vanden Abeele, F.; Bidaux, G.; Gordienko, D.; Beck, B.; Panchin, Y.V.; Baranova, A.V.; Ivanov, D.V.; Skryma, R.; Prevarskaya, N. Functional implications of calcium permeability of the channel formed by pannexin 1. J. Cell Biol. 2006, 174, 535–546, doi:10.1083/jcb.200601115.
- Kim, J.C.; Son, M.J.; Woo, S.H. Ca2+ Signaling triggered by shear-autocrine P2X receptor pathway in rat atrial myocytes. Cell. Physiol. Biochem. 2018, 50, 2296–2313, doi:10.1159/000495089
- Cogliati, B.; Mennecier, G.; Willebrords, J.; Da Silva, T.C.; Maes, M.; Pereira, I.V.A.; Crespo Yanguas, S.; Hernandez-Blazquez, F.J.; Dagli, M.L.Z.; Vinken, M. Connexins, Pannexins, and Their Channels in Fibroproliferative Diseases. J. Membr. Biol. 2016, 249, 199–213, doi:10.1007/s00232-016-9881-6.
- Tribulova, N.; Szeiffova Bacova, B.; Benova, T.; Viczenczova, C. Can we protect from malignant arrhythmias by modulation of cardiac cell-to-cell coupling? J. Electrocardiol. 2015, 48, 434–440, doi:10.1016/j.jelectrocard.2015.02.006.
- Baum, J.R.; Dolmatova, E.; Tan, A.; Duffy, H.S. Omega 3 fatty acid inhibition of inflammatory cytokine-mediated Connexin43 regulation in the heart. Front. Physiol. 2012, 3, 272, doi:10.3389/fphys.2012.00272.
- Egan Benova, T.; Szeiffova Bacova, B.; Viczenczova, C.; Diez, E.; Barancik, M.; Tribulova, N. Protection of cardiac cell-to-cell coupling attenuate myocardial remodeling and proarrhythmia induced by hypertension. Physiol. Res. 2016, 65, S29–S42, doi:10.33549/physiolres.933391.
- Rivaud, M.R.; Delmar, M.; Remme, C.A. Heritable arrhythmia syndromes associated with abnormal cardiac sodium channel function: Ionic and non-ionic mechanisms. Cardiovasc. Res. 2020, 116, 1557–1570, doi:10.1093/cvr/cvaa082.
- van der Voorn, S.M.; Te Riele, A.S.J.M.; Basso, C.; Calkins, H.; Remme, C.A.; van Veen, T.A.B. Arrhythmogenic cardiomyopathy: Pathogenesis, pro-arrhythmic remodelling, and novel approaches for risk stratification and therapy. Cardiovasc. Res. 2020, 116, 1571–1584, doi:10.1093/cvr/cvaa084.
- Chatterjee, D.; Pieroni, M.; Fatah, M.; Charpentier, F.; Cunningham, K.S.; Spears, D.A.; Chatterjee, D.; Suna, G.; Martjin Bos, J.; Ackerman, M.J.; et al. An autoantibody profile detects Brugada syndrome and identifies abnormally expressed myocardial proteins. Eur. Heart J. 2020, 41, 2878-2890A, doi:10.1093/eurheartj/ehaa383.
- Sáez, J.C.; Green, C. Involvement of connexin hemichannels in the inflammatory response of chronic diseases. Int. J. Mol. Sci. 2018, 19, 1–3, doi:10.3390/ijms19092469
- Burnstock, G. Pathophysiology and Therapeutic Potential of Purinergic Signaling. Pharmacol. Rev. 2006, 58, 58–86, doi:10.1124/pr.58.1.5.58.
- Billaud, M.; Sandilos, J.K.; Isakson, B.E. Pannexin 1 in the Regulation of Vascular Tone. Trends Cardiovasc. Med. 2012, 22, 68–72, doi:10.1016/j.tcm.2012.06.014.
- Burnstock, G.; Pelleg, A. Cardiac purinergic signalling in health and disease. Purinergic Signal. 2015, 11, 1–46, doi:10.1007/s11302-014-9436-1.
- Boengler, K.; Dodoni, G.; Rodriguez-Sinovas, A.; Cabestrero, A.; Ruiz-Meana, M.; Gres, P.; Konietzka, I.; Lopez-Iglesias, C.; Garcia-Dorado, D.; Di Lisa, F.; et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc. Res. 2005, 67, 234–244, doi:10.1016/j.cardiores.2005.04.014.
- Tribulová, N.; Knezl, V.; Okruhlicová, L.; Slezák, J. Myocardial gap junctions: Targets for novel approaches in the prevention of life-threatening cardiac arrhythmias. Physiol. Res. 2008, 57, 1–13.
- Tribulova, N.; Seki, S.; Radosinska, J.; Kaplan, P.; Babusikova, E.; Knezl, V.; Mochizuki, S. Myocardial Ca2+ handling and cell-to-cell coupling, key factors in prevention of sudden cardiac death1. Can. J. Physiol. Pharmacol. 2009, 87, 1120–1129, doi:10.1139/Y09-106.
- Tribulova, N.; Egan Benova, T.; Szeiffova Bacova, B.; Viczenczova, C.; Barancik, M. New aspects of pathogenesis of atrial fibrillation: Remodeling of intercalated discs. J. Physiol. Pharmacol. 2015, 66, 625–634.
- Tribulova, N.; Bacova, B.S.; Benova, T.E.; Knezl, V.; Barancik, M.; Slezak, J. Omega-3 index and anti-arrhythmic potential of omega-3 PUFAs. Nutrients 2017, 9, 1–21, doi:10.3390/nu9111191.
- Prado, N.J.; Egan Beňová, T.; Diez, E.R.; Knezl, V.; Lipták, B.; Ponce Zumino, A.Z.; Llamedo-Soria, M.; Szeiffová Bačová, B.; Miatello, R.M.; Tribulová, N. Melatonin receptor activation protects against low potassium-induced ventricular fibrillation by preserving action potentials and connexin-43 topology in isolated rat hearts. J. Pineal Res. 2019, 67, 1–14, doi:10.1111/jpi.12605.
- Nagibin, V.; Egan Benova, T.; Viczenczova, C.; Szeiffova Bacova, B.; Dovinova, I.; Barancik, M.; Tribulova, N. Ageing related down-regulation of myocardial connexin-43 and up-regulation of MMP-2 may predict propensity to atrial fibrillation in experimental animals. Physiol. Res. 2016, 65, S91–S100, doi:10.33549/physiolres.933389.
- Yanguas, S.C.; da Silva, T.C.; Pereira, I.V.A.; Willebrords, J.; Maes, M.; Nogueira, M.S.; de Castro, I.A.; Leclercq, I.; Romualdo, G.R.; Barbisan, L.F.; et al. TAT-Gap19 and Carbenoxolone alleviate liver fibrosis in mice. Int. J. Mol. Sci. 2018, 19, doi:10.3390/ijms19030817.
- Crespo Yanguas, S.; Willebrords, J.; Johnstone, S.R.; Maes, M.; Decrock, E.; De Bock, M.; Leybaert, L.; Cogliati, B.; Vinken, M. Pannexin1 as mediator of inflammation and cell death. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 51–61, doi:10.1016/j.bbamcr.2016.10.006.
- Ongstad, E.L.; Gourdie, R.G. Can heart function lost to disease be regenerated by therapeutic targeting of cardiac scar tissue? Semin. Cell Dev. Biol. 2016, 58, 41–54, doi:10.1016/j.semcdb.2016.05.020.
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867, doi:10.1038/nature09413.
- Adamson, S.E.; Leitinger, N. The role of pannexin1 in the induction and resolution of inflammation. FEBS Lett. 2014, 588, 1416–1422, doi:10.1016/j.febslet.2014.03.009.
- Qu, Y.; Misaghi, S.; Newton, K.; Gilmour, L.L.; Louie, S.; Cupp, J.E.; Dubyak, G.R.; Hackos, D.; Dixit, V.M. Pannexin-1 Is Required for ATP Release during Apoptosis but Not for Inflammasome Activation. J. Immunol. 2011, 186, 6553–6561, doi:10.4049/jimmunol.1100478.
- Maslieieva, V.; Thompson, R.J. A critical role for pannexin-1 in activation of innate immune cells of the choroid plexus. Channels 2014, 8, 131–141, doi:10.4161/chan.27653.
- Chelko, S.P.; Asimaki, A.; Lowenthal, J.; Bueno-Beti, C.; Bedja, D.; Scalco, A.; Amat-Alarcon, N.; Andersen, P.; Judge, D.P.; Tung, L.; et al. Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy. Circulation 2019, 140, 1491–1505, doi:10.1161/CIRCULATIONAHA.119.040676.
- Fridolfsson, H.N.; Roth, D.M.; Insel, P.A.; Patel, H.H. Regulation of intracellular signaling and function by caveolin. FASEB J. 2014, 28, 3823–3831, doi:10.1096/fj.14-252320.
- Head, B.P.; Patel, H.H.; Insel, P.A. Interaction of membrane/lipid rafts with the cytoskeleton: Impact on signaling and function: Membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim. Biophys. Acta Biomembr. 2014, 1838, 532–545, doi:10.1016/j.bbamem.2013.07.018.
- Cascio, M. Connexins and their environment: Effects of lipids composition on ion channels. Biochim. Biophys. Acta Biomembr. 2005, 1711, 142–153, doi:10.1016/j.bbamem.2004.12.001.
- Chung, T.H.; Wang, S.M.; Liang, J.Y.; Yang, S.H.; Wu, J.C. The interaction of estrogen receptor α and caveolin-3 regulates connexin43 phosphorylation in metabolic inhibition-treated rat cardiomyocytes. Int. J. Biochem. Cell Biol. 2009, 41, 2323–2333, doi:10.1016/j.biocel.2009.06.001.
- Liu, L.; Li, Y.; Lin, J.; Liang, Q.; Sheng, X.; Wu, J.; Huang, R.; Liu, S.; Li, Y. Connexin43 interacts with Caveolin-3 in the heart. Mol. Biol. Rep. 2010, 37, 1685–1691, doi:10.1007/s11033-009-9584-5.
- Yang, K.C.; Rutledge, C.A.; Mao, M.; Bakhshi, F.R.; Xie, A.; Liu, H.; Bonini, M.G.; Patel, H.H.; Minshall, R.D.; Dudley, S.C. Caveolin-1 modulates cardiac gap junction homeostasis and arrhythmogenecity by regulating csrc tyrosine kinase. Circ. Arrhythmia Electrophysiol. 2014, 7, 701–710, doi:10.1161/CIRCEP.113.001394.
- Saliez, J.; Bouzin, C.; Rath, G.; Ghisdal, P.; Desjardins, F.; Rezzani, R.; Rodella, L.F.; Vriens, J.; Nilius, B.; Feron, O.; et al. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation-Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 2008, 117, 1065–1074, doi:10.1161/CIRCULATIONAHA.107.731679.
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39, doi:10.1038/35036052.
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.L. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006, 47, 74–80, doi:10.1161/10.1161/01.HYP.0000196727.53300.62.
- Maguy, A.; Hebert, T.E.; Nattel, S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc. Res. 2006, 69, 798–807, doi:10.1016/j.cardiores.2005.11.013.
- Tsutsumi, Y.M.; Horikawa, Y.T.; Jennings, M.M.; Kidd, M.W.; Niesman, I.R.; Yokoyama, U.; Head, B.P.; Hagiwara, Y.; Ishikawa, Y.; Miyanohara, A.; et al. Cardiac-specific overexpression of caveolin-3 induces endogenous cardiac protection by mimicking ischemic preconditioning. Circulation 2008, 118, 1979–1988, doi:10.1161/CIRCULATIONAHA.108.788331.
- Rath, G.; Dessy, C.; Feron, O. Caveolae, Caveolin and control of vascular tone: Nitric oxide (NO) and endothelium derived hyperpolarizing factor (EDHF) regulation. J. Physiol. Pharmacol. 2009, 60, 105–109.
- Horikawa, Y.T.; Panneerselvam, M.; Kawaraguchi, Y.; Tsutsumi, Y.M.; Ali, S.S.; Balijepalli, R.C.; Murray, F.; Head, B.P.; Niesman, I.R.; Rieg, T.; et al. Cardiac-specific overexpression of caveolin-3 attenuates cardiac hypertrophy and increases natriuretic peptide expression and signaling. J. Am. Coll. Cardiol. 2011, 57, 2273–2283, doi:10.1016/j.jacc.2010.12.032.
- Jansen, J.A.; Noorman, M.; Musa, H.; Stein, M.; De Jong, S.; Van Der Nagel, R.; Hund, T.J.; Mohler, P.J.; Vos, M.A.; Van Veen, T.A.; et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Hear. Rhythm 2012, 9, 600–607, doi:10.1016/j.hrthm.2011.11.025.
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K.I. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Hear. Circ. Physiol. 2007, 293, 1714–1720, doi:10.1152/ajpheart.00022.2007.
- Sáez, J.C.; Schalper, K.A.; Retamal, M.A.; Orellana, J.A.; Shoji, K.F.; Bennett, M.V.L. Cell membrane permeabilization via connexin hemichannels in living and dying cells. Exp. Cell Res. 2010, 316, 2377–2389, doi:10.1016/j.yexcr.2010.05.026.
- Ongstad, E.; Kohl, P. Fibroblast-myocyte coupling in the heart: Potential relevance for therapeutic interventions. J. Mol. Cell. Cardiol. 2016, 91, 238–246, doi:10.1016/j.yjmcc.2016.01.010.
- John, S.A.; Kondo, R.; Wang, S.Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240, doi:10.1074/jbc.274.1.236.
- Jansen, J.A.; Noorman, M.; Musa, H.; Stein, M.; Jong, S. De; Der, R. Van; Hund, T.J.; Mohler, P.J.; Vos, M.A.; Veen, T.A. Van; et al. NIH Public Access. 2013, 9, 600–607, doi:10.1016/j.hrthm.2011.11.025.Reduced.
- Decrock, E.; Vinken, M.; De Vuyst, E.; Krysko, D.V.; D’Herde, K.; Vanhaecke, T.; Vandenabeele, P.; Rogiers, V.; Leybaert, L. Connexin-related signaling in cell death: To live or let die? Cell Death Differ. 2009, 16, 524–536, doi:10.1038/cdd.2008.196.
- Connexin-related signaling in cell death: To live or let die? Cell Death Differ. 2009, 16, 524–536, doi:10.1038/cdd.2008.196.
- Li, F.; Sugishita, K.; Su, Z.; Ueda, I.; Barry, W.H. Activation of connexin-43 hemichannels can elevate [Ca2+]i and[Na+]i in rabbit ventricular myocytes during metabolic inhibition. J. Mol. Cell. Cardiol. 2001, 33, 2145–2155, doi:10.1006/jmcc.2001.1477.
- De Bock, M.; Wang, N.; Bol, M.; Decrock, E.; Ponsaerts, R.; Bultynck, G.; Dupont, G.; Leybaert, L. Connexin 43 hemichannels contribute to cytoplasmic Ca2+ oscillations by providing a bimodal Ca2+-dependent Ca2+ entry pathway. J. Biol. Chem. 2012, 287, 12250–12266, doi:10.1074/jbc.M111.299610.
- Soetkamp, D.; Nguyen, T.T.; Menazza, S.; Hirschhäuser, C.; Hendgen-Cotta, U.B.; Rassaf, T.; Schlüter, K.D.; Boengler, K.; Murphy, E.; Schulz, R. S-nitrosation of mitochondrial connexin 43 regulates mitochondrial function. Basic Res. Cardiol. 2014, 109, 433, doi:10.1007/s00395-014-0433-x.
- Lillo, M.A.; Himelman, E.; Shirokova, N.; Xie, L.H.; Fraidenraich, D.; Contreras, J.E. S-nitrosylation of connexin43 hemichannels elicits cardiac stress–induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight 2019, 4, e130091, doi:10.1172/jci.insight.130091.
- Retamal, M.A.; Cortés, C.J.; Reuss, L.; Bennett, M.V.L.; Sáez, J.C. S-nitrosylation and permeation through connexin 43 hemichannels in astrocytes: Induction by oxidant stress and reversal by reducing agents. Proc. Natl. Acad. Sci. USA 2006, 103, 4475–4480, doi:10.1073/pnas.0511118103.
- Lohman, A.W.; Weaver, J.L.; Billaud, M.; Sandilos, J.K.; Griffiths, R.; Straub, A.C.; Penuela, S.; Leitinger, N.; Laird, D.W.; Bayliss, D.A.; et al. S-nitrosylation inhibits pannexin 1 channel function. J. Biol. Chem. 2012, 287, 39602–39612, doi:10.1074/jbc.M112.397976.
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Bennett, M.V.L.; Sáez, J.C. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc. Natl. Acad. Sci. USA 2007, 104, 8322–8327, doi:10.1073/pnas.0702456104.
- Song, Y.; Belardinelli, L. ATP promotes development of afterdepolarizations and triggered activity in cardiac myocytes. Am. J. Physiol. Hear. Circ. Physiol. 1994, 267, 2005–2011, doi:10.1152/ajpheart.1994.267.5.h2005.
- Zhang, B.-X.; Desnoyer, R.W.; Bond, M. Extracellular Adenosine Triphosphate Triggers Arrhythmias and Elemental Redistribution in Electrically Stimulated Rat Cardiac Myocytes. Microsc. Microanal. 2001, 7, 48–55, doi:10.1007/s100050010056.
- Gurung, I.S.; Kalin, A.; Grace, A.A.; Huang, C.L.H. Activation of purinergic receptors by ATP induces ventricular tachycardia by membrane depolarization and modifications of Ca2+ homeostasis. J. Mol. Cell. Cardiol. 2009, 47, 622–633, doi:10.1016/j.yjmcc.2009.08.001.
- Pustovit, K.B.; Potekhina, V.M.; Ivanova, A.D.; Petrov, A.M.; Abramochkin, D.V.; Kuzmin, V.S. Extracellular ATP and β-NAD alter electrical properties and cholinergic effects in the rat heart in age-specific manner. Purinergic Signal. 2019, 15, 107–117, doi:10.1007/s11302-019-09645-6.
- Nattel, S.; Maguy, A.; Le Bouter, S.; Yeh, Y.H. Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiol. Rev. 2007, 87, 425–456, doi:10.1152/physrev.00014.2006.
- Jiang, C.Y.; Jiang, R.H.; Matsuo, S.; Fu, G.S. ATP revealed extra pulmonary vein source of atrial fibrillation after circumferential pulmonary vein isolation. PACE Pacing Clin. Electrophysiol. 2010, 33, 248–251, doi:10.1111/j.1540-8159.2009.02532.x.
- Miyazaki, S.; Taniguchi, H.; Komatsu, Y.; Uchiyama, T.; Kusa, S.; Nakamura, H.; Hachiya, H.; Hirao, K.; Iesaka, Y. Clinical impact of adenosine triphosphate injection on arrhythmogenic superior vena cava in the context of atrial fibrillation ablation. Circ. Arrhythmia Electrophysiol. 2013, 6, 497–503, doi:10.1161/CIRCEP.113.000281.
- Cheung, J.W.; Lin, F.S.; Ip, J.E.; Bender, S.R.; Siddiqi, F.K.; Liu, C.F.; Thomas, G.; Markowitz, S.M.; Lerman, B.B. Adenosine-induced pulmonary vein ectopy as a predictor of recurrent atrial fibrillation after pulmonary vein isolation. Circ. Arrhythmia Electrophysiol. 2013, 6, 1066–1073, doi:10.1161/CIRCEP.113.000796.
- McLellan, A.J.A.; Kumar, S.; Smith, C.; Morton, J.B.; Kalman, J.M.; Kistler, P.M. The role of adenosine following pulmonary vein isolation in patients undergoing catheter ablation for atrial fibrillation: A systematic review. J. Cardiovasc. Electrophysiol. 2013, 24, 742–751, doi:10.1111/jce.12121.
- Ip, J.E.; Cheung, J.W.; Chung, J.H.; Liu, C.F.; Thomas, G.; Markowitz, S.M.; Lerman, B.B. Adenosine-induced atrial fibrillation insights into mechanism. Circ. Arrhythmia Electrophysiol. 2013, 6, 34–37, doi:10.1161/CIRCEP.113.000480.
- Maille, B.; Marlinge, M.; Vairo, D.; Mottola, G.; Koutbi, L.; Deharo, P.; Gastaldi, M.; Gaudry, M.; Guiol, C.; Bottone, S.; et al. Adenosine plasma level in patients with paroxysmal or persistent atrial fibrillation and normal heart during ablation procedure and/or cardioversion. Purinergic Signal. 2019, 15, 45–52, doi:10.1007/s11302-018-9636-1.
- Mahoney, V.M.; Mezzano, V.; Mirams, G.R.; Maass, K.; Li, Z.; Cerrone, M.; Vasquez, C.; Bapat, A.; Delmar, M.; Morley, G.E. Connexin43 contributes to electrotonic conduction across scar tissue in the intact heart. Sci. Rep. 2016, 6, 26744, doi:10.1038/srep26744.
- Johnson, R.D.; Camelliti, P. Role of non-myocyte gap junctions and connexin hemichannels in cardiovascular health and disease: Novel therapeutic targets? Int. J. Mol. Sci. 2018, 19, 866, doi:10.3390/ijms19030866.
- Vasquez, C.; Benamer, N.; Morley, G.E. The cardiac fibroblast: Functional and electrophysiological considerations in healthy and diseased hearts. J. Cardiovasc. Pharmacol. 2011, 57, 380–388, doi:10.1097/FJC.0b013e31820cda19.
- Zhang, Y.; Kanter, E.M.; Yamada, K.A. Remodeling of cardiac fibroblasts following myocardial infarction results in increased gap junction intercellular communication. Cardiovasc. Pathol. 2010, 19, e233–e240, doi:10.1016/j.carpath.2009.12.002.
- Banerjee, I.; Yekkala, K.; Borg, T.K.; Baudino, T.A. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N. Y. Acad. Sci. 2006, 1080, 76–84, doi:10.1196/annals.1380.007.
- Kohl, P.; Gourdie, R.G. Fibroblast-myocyte electrotonic coupling: Does it occur in native cardiac tissue? J. Mol. Cell. Cardiol. 2014, 70, 37–46, doi:10.1016/j.yjmcc.2013.12.024.
- Mayourian, J.; Ceholski, D.K.; Gonzalez, D.M.; Cashman, T.J.; Sahoo, S.; Hajjar, R.J.; Costa, K.D. Physiologic, Pathologic, and Therapeutic Paracrine Modulation of Cardiac Excitation-Contraction Coupling. Circ. Res. 2018, 122, 167–183, doi:10.1161/CIRCRESAHA.117.311589.
- Banerjee, I.; Yekkala, K.; Borg, T.K.; Baudino, T.A. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N. Y. Acad. Sci. 2006, 1080, 76–84, doi:10.1196/annals.1380.007.
- Kohl, P.; Gourdie, R.G. Fibroblast-myocyte electrotonic coupling: Does it occur in native cardiac tissue? J. Mol. Cell. Cardiol. 2014, 70, 37–46, doi:10.1016/j.yjmcc.2013.12.024.
- Mayourian, J.; Ceholski, D.K.; Gonzalez, D.M.; Cashman, T.J.; Sahoo, S.; Hajjar, R.J.; Costa, K.D. Physiologic, Pathologic, and Therapeutic Paracrine Modulation of Cardiac Excitation-Contraction Coupling. Circ. Res. 2018, 122, 167–183, doi:10.1161/CIRCRESAHA.117.311589.