Peripartum cardiomyopathy (PPCM) is a condition in which heart failure and left ventricular systolic dysfunction occur in the last month of pregnancy or within months following delivery.
- peripartum cardiomyopathy
- genetic cardiomyopathy
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Peripartum cardiomyopathy (PPCM) is a rare form of cardiac muscle disease associated with pregnancy. PPCM is a potentially lethal condition which is defined as heart failure (HF) and left ventricular systolic dysfunction occurring late in pregnancy or within months following delivery and in the absence of other causes of HF [1]. Although left ventricular recovery is possible up to 12 months following diagnosis, adverse outcomes such as irreversible HF, arrhythmia and sudden cardiac death are frequent [2,3].
The reported incidence of PPCM varies greatly between population groups, from approximately 1:1000 pregnancies in South Africa [4] to 1:10,000 pregnancies in Denmark [5]. Higher incidence of PPCM in Nigeria, South Africa and African Americans reported historically suggests that individuals of African descent may be at greater risk of PPCM [4,6]. However, a multicentre registry spanning 43 countries indicated that PPCM is a global disease that is likely to be under-reported in many instances and can occur in women of any ethnic background [7]. Other documented PPCM risk factors include multiparity, increased maternal age, and family history of cardiovascular disease [1]. The disparity in PPCM prevalence across various populations suggests that apart from environmental factors, a strong genetic basis for the condition may exist.
Precisely how PPCM develops is unclear, although there are several proposed mechanisms (Figure 1). It has been observed that pregnancy induces dramatic haemodynamic changes, such as reduced afterload and an increase in cardiac output and blood volume [8]. These changes trigger homeostatic and structural remodelling of cardiovascular tissues which results in exacerbated cardiac stress. As such, the hormonal changes associated with parturition may trigger endothelial dysfunction and PPCM in susceptible women [9]. Over the last decade, much progress has been made towards understanding the genetics of familial forms of cardiomyopathy such as dilated cardiomyopathy (DCM). Given the role of family history and genetic factors in PPCM, these advances have afforded opportunities to better understand the genetics of PPCM. This review summarises current knowledge of the genetic contribution to PPCM and highlights potential avenues for future research.
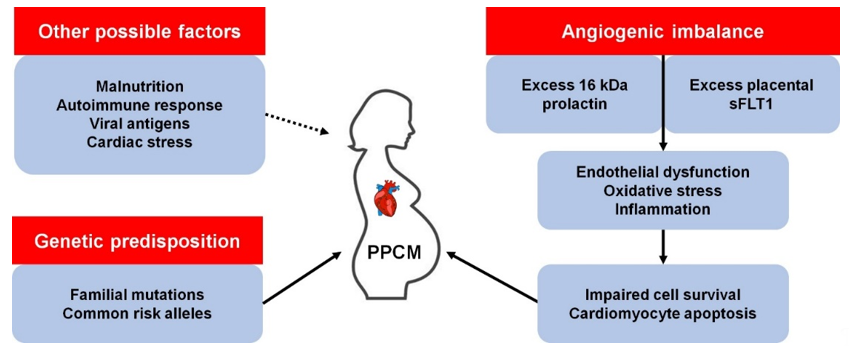
Figure 1. Known and possible mechanisms of peripartum cardiomyopathy (PPCM) pathophysiology. kDa, kilodalton; sFLT1, soluble fms-like tyrosine kinase-1.
2. The Role of Familial Cardiomyopathy Genes in PPCM
One of the first indications of the role of genetic susceptibility in PPCM pathogenesis was familial clustering of PPCM with other forms of cardiomyopathy. Three instances of familial PPCM were reported in 1963 by Pierce et al. [10]; many similar familial occurrences of PPCM and DCM have been subsequently documented [11–17]. Although PPCM is a distinct clinical entity, these observations of overlap with familial DCM indicate that, in at least a subset of patients, PPCM may form part of the clinical and genetic spectrum of DCM. DCM itself is a vastly heterogeneous disease, genetically overlapping with hypertrophic cardiomyopathy (HCM), arrhythmogenic cardiomyopathy (ACM), and channelopathies: mutations in many of these genes have also been described in PPCM patients (Table 1; Figure 2).
Table 1. Summary of genes associated with PPCM to date.
|
Gene |
Molecular Function |
Mutation Types in PPCM |
Other Associated Disorders |
|
BAG3 |
Co-chaperone, Z disk |
Truncating |
DCM, MFM |
|
DMD |
Sarcolemma, structure |
Truncating |
DCM, MD |
|
DSP |
Desmosome, cell–cell adhesion |
Truncating |
ACM, DCM, keratodermas |
|
FKTN |
May process dystrophin |
Truncating/missense |
DCM, MD |
|
GNB3 |
G protein subunit |
Association with outcome |
Hypertension, night blindness |
|
KCNH2 |
K+ channel, cardiac conduction |
Truncating |
Long QT syndrome |
|
LAMP2 |
Lysosome, autophagy |
Truncating/missense |
Danon disease, DCM, HCM |
|
LMNA |
Nuclear lamina, structure |
Truncating |
DCM, MD |
|
MYBPC3 |
Sarcomere, cardiac contraction |
Missense |
DCM, HCM, LVNC |
|
MYH6 |
Sarcomere, cardiac contraction |
Truncating/missense |
CHD, DCM, HCM |
|
MYH7 |
Sarcomere, cardiac contraction |
Missense |
DCM, HCM, LVNC, MD |
|
PSEN2 |
May regulate APP processing |
Missense |
Alzheimer’s disease, DCM |
|
PTHLH |
Hormone |
Association with risk |
Brachydactyly |
|
RET |
Protooncogene |
Missense |
Multiple endocrine neoplasia |
|
SCN5A |
NA+ channel, cardiac conduction |
Missense |
AF, DCM, Long QT syndrome, VF |
|
SYNM |
Cytoskeleton |
Truncating |
- |
|
TNNC1 |
Sarcomere, cardiac contraction |
Missense |
DCM, HCM |
|
TNNT2 |
Sarcomere, cardiac contraction |
Missense |
DCM, HCM, LVNC, RCM |
|
TPM1 |
Sarcomere, cardiac contraction |
Truncating |
DCM, HCM, LVNC |
|
TTN |
Sarcomere, cardiac contraction |
Truncating |
DCM, HCM, MD, MFM |
|
VCL |
Cytoskeleton |
Truncating |
DCM, HCM |
ACM, arrhythmogenic cardiomyopathy; AF, atrial fibrillation; APP, amyloid precursor protein; CHD, congenital heart disease; DCM, dilated cardiomyopathy, HCM, hypertrophic cardiomyopathy; HSP, heat shock protein; LVNC, left ventricular noncompaction; MD, muscular dystrophy; MFM, myofibrillar myopathy; RCM, restrictive cardiomyopathy; VF, ventricular fibrillation.
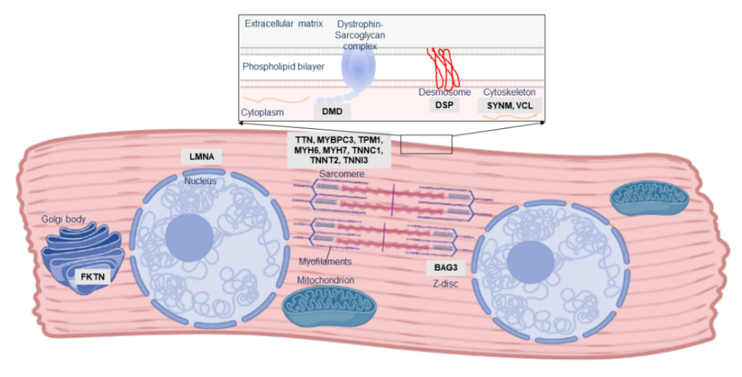
Figure 2. Localisation and function of PPCM-associated genes in the cardiomyocyte. The majority of PPCM genes, including Titin (TTN), comprise the cardiac sarcomere, but mutations affecting numerous other aspects of cardiomyocyte function have been implicated in PPCM pathogenesis.
In what appears to be the first documented genetic cause of PPCM, a 2001 case report described a female carrier of a Duchenne Muscular Dystrophy-causing DMD mutation who developed PPCM during the 36th week of pregnancy [18]. As an X-linked gene, most female carriers of DMD mutations are asymptomatic although some may develop mild disease or cardiomyopathy, and it was unclear if the PPCM, in this case, was related to her carrier status. However, similar case reports of PPCM amongst DMD and LAMP2 X-linked mutation carriers have been described [19–21]. LAMP2 mutations cause Danon disease, a form of cardiomyopathy and skeletal myopathy, through impairment of macroautophagy [22], while DMD mutations cause muscular dystrophy and/or DCM through the loss of the stabilising protein dystrophin, leaving myocytes vulnerable to oxidative stress and calcium overload [23]. In both cases, it is thought that skewed X-chromosome inactivation, in which only some cells express the DMD or LAMP2 mutation, underlies the comparatively milder cardiac phenotype observed in females [24]. It is possible, too, that this mosaic status in females may be exacerbated by stress conditions such as pregnancy, although no evidence of this has been reported as yet.
DMD and LAMP2 mutations are very rare causes of DCM; notably, most known DCM-causing mutations affect the sarcomere, causing cardiomyopathy through impaired force generation or reduced contractility in the heart. Early investigations reported several mutations in the sarcomeric genes MYBPC3, MYH6, MYH7, TNNC1 and TNNT2 in Dutch and American PPCM cohorts [13,17,25]. As such, similar studies across several other populations may provide critical information into the role of these mutations in PPCM.
As knowledge of DCM genetics has evolved over time, so too has knowledge of the genetic contribution to PPCM. Subsequent to these initial investigations, the giant sarcomeric protein-coding gene TTN emerged as a key gene in cardiomyopathy, accounting for up to 25% of familial DCM cases [26]. As the second-longest gene in humans, genetic variation in TTN is common [27], and it may be challenging to differentiate benign polymorphisms from true cardiomyopathy-causing mutations. However, frameshift insertions/deletions and nonsense mutations (truncating mutations) in TTN have been reported at an increased prevalence in DCM patients [26,27]. The mechanisms underlying TTN cardiomyopathy are thought to involve impairment of force generation or transmission in the myocardium or disrupted signalling [28]. BAG3 has also recently emerged as a major cardiomyopathy gene [29]. BAG3 encodes a multifunction co-chaperone protein involved in myocardial protein homeostasis, stabilisation of the myocardial Z disk, and modulation of cardiac contraction [30]. Mutations in BAG3 can cause cardiomyopathy through impairments in any of the protein’s diverse functions [31].
The emergence of high throughput next-generation sequencing (NGS) platforms enabled the rapid sequencing of patient genes and has highlighted the genetic overlap between different forms of heritable cardiomyopathy. Following these genetic advances, NGS studies of PPCM cohorts have revealed truncating mutations in TTN as the predominant genetic contributor to PPCM in American, Australian and European patients [32–34]. Although such TTN mutations are present in healthy individuals, they were found to be significantly more prevalent amongst PPCM patients [33]. In addition, the TTN mutations were mostly localised to the protein domains already associated with DCM (A-band), thereby implicating their possible role in PPCM [33]. In these NGS studies, mutations were reported in other cardiac disease genes, including disruptions of BAG3 [32–34]. Cumulatively, these cohort studies of PPCM have identified pathogenic mutations in 23% of cases (Figure 3). Studies of isolated PPCM cases have further implicated TTN in severe PPCM [35], as well as other disease genes such as KCNH2, RET and TXNRD2 [16,36,37], although the contribution of these genes to PPCM is unclear: the mutations in these cases may merely reflect the co-occurrence of genetic disorders with PPCM. Mutations in FKTN, RBM20, LMNA and DSP were also linked to PPCM through large cardiomyopathy family studies [38–41].
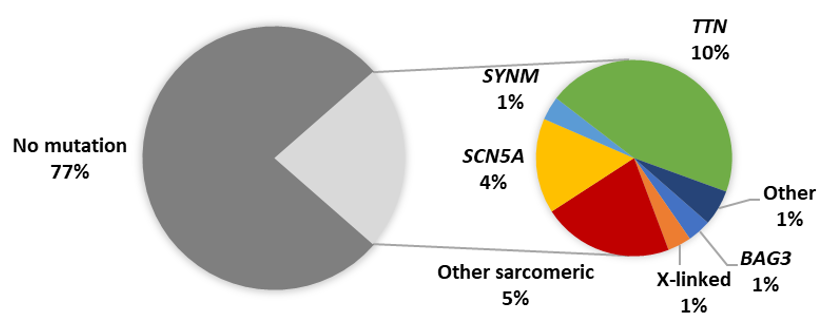
Figure 3. Proportion of reported PPCM patients with pathogenic mutations. Other sarcomeric genes: MYBPC3, MYH6, MYH7, TNNC1, TNNT2, TPM1; X-linked genes: DMD, LAMP2; Other genes: DSP, PSEN2, VCL.
The diverse genetic aetiology of PPCM (Table 1) mirrors the genetic profile of DCM and strengthens the notion that PPCM may share genetic determinants with familial cardiomyopathy. In PPCM, as in DCM, a high proportion of TTN truncating variants is observed, and at present appears to be the greatest single genetic contributor to disease pathogenesis [32,33]. The mutation detection rate of genetic testing in DCM ranges from 15% to 40%, with familial history and larger gene panels contributing to higher mutation yields [42]. Although within this range, the relatively low yield (~20%) in PPCM may be attributed to the substantially fewer patients who have been screened so far, combined with fewer genes being sequenced in earlier studies.
When considering the worldwide distribution of PPCM-associated mutations (Figure 4), it is noteworthy that the majority have been identified in American and European populations, but even then, many of the genes are limited to single observations and case reports. Analysis of participants of African descent by Ware et al. [33] revealed mutation yields that were akin to those in individuals of European ancestry, indicating a similar genetic basis of PPCM in different population groups. This also suggests that mutation screening of African PPCM cohorts may be warranted. In DCM, the clinical utility of genetic testing can extend to family management and individual treatment and prognosis, through the characterisation of genotype-phenotype correlations. For example, LMNA mutations are associated with severe DCM with high penetrance and increased risk of sudden death, while TTN mutations have been associated with milder disease with reduced penetrance [43]. Mutations in these genes have been reported in PPCM, but their implications on phenotype and outcome are still to be determined. Further research is also needed to determine whether the co-occurrence of cardiomyopathy mutations in PPCM patients reflects causality or if, rather, it is the physiological strain of pregnancy that reveals cardiomyopathy in previously asymptomatic mutation carriers.
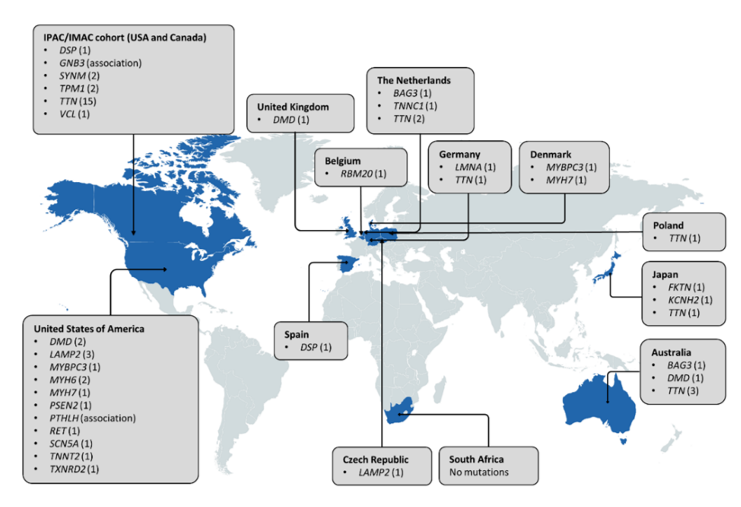
Figure 4. Distribution of genes associated with PPCM to date. Countries in which genetic investigations of PPCM patient(s) have been conducted are indicated in blue. The number of pathogenic mutations identified in each gene is indicated in parentheses. Three pathogenic MYH7 mutations and seven SCN5A mutations are not included as the countries of origin could not be determined.
References
- Bauersachs, J.; Konig, T.; van der Meer, P.; Petrie, M.C.; Hilfiker-Kleiner, D.; Mbakwem, A. Pathophysiology, diagnosis and management of peripartum cardiomyopathy: A position statement from the Heart Failure Association of the European Society of Cardiology Study Group on peripartum cardiomyopathy. J. Heart Fail 2019, 21, 827–843, doi:10.1002/ejhf.1493.
- Hoevelmann, J.; Hahnle, L.; Hahnle, J.; Sliwa, K.; Viljoen, C. Detection and management of arrhythmias in peripartum cardiomyopathy. Diagn. Ther. 2020, 10, 325–335, doi:10.21037/cdt.2019.05.03.
- McNamara, D.M.; Elkayam, U.; Alharethi, R.; Damp, J.; Hsich, E.; Ewald, G. Clinical outcomes for peripartum cardiomyopathy in North America: Results of the IPAC study (Investigations of Pregnancy-Associated Cardiomyopathy). Am. Coll. Cardiol. 2015, 66, 905–914, doi:10.1016/j.jacc.2015.06.1309.
- Desai, D.; Moodley, J.; Naidoo, D. Peripartum cardiomyopathy: Experiences at King Edward VIII Hospital, Durban, South Africa and a review of the literature. Doct. 1995, 25, 118–123, doi:10.1177/004947559502500310.
- Ersbøll, A.S.; Johansen, M.; Damm, P.; Rasmussen, S.; Vejlstrup, N.G.; Gustafsson, F. Peripartum cardiomyopathy in Denmark: A retrospective, population-based study of incidence, management and outcome. J. Heart Fail 2017, 19, 1712–1720, doi:10.1002/ejhf.882.
- Kolte, D.; Khera, S.; Aronow, W.S.; Palaniswamy, C.; Mujib, M.; Ahn, C. Temporal trends in incidence and outcomes of peripartum cardiomyopathy in the United States: A nationwide population-based study. Am. Heart Assoc. 2014, 3, e001056, doi:10.1161/JAHA.114.001056.
- Sliwa, K.; Mebazaa, A.; Hilfiker-Kleiner, D.; Petrie, M.C.; Maggioni, A.P.; Laroche, C. Clinical characteristics of patients from the worldwide registry on peripartum cardiomyopathy (PPCM): EURObservational Research Programme in conjunction with the Heart Failure Association of the European Society of Cardiology Study Group on PPCM. J. Heart Fail 2017, 19, 1131–1141, doi:10.1002/ejhf.780.
- Hall, M.E.; George, E.M.; Granger, J.P. The heart during pregnancy. Esp. Cardiol. 2011, 64, 1045–1050, doi:10.1016/j.recesp.2011.07.009.
- Hilfiker-Kleiner, D.; Kaminski, K.; Podewski, E.; Bonda, T.; Schaefer, A.; Sliwa, K. A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 2007, 128, 589–600, doi:10.1016/j.cell.2006.12.036.
- Pierce, J.A.; Price, B.O.; Joyce, J.W. Familial occurrence of postpartal heart failure. Arch Intern Med 1963, 111, 651–655, doi:10.1001/archinte.1963.03620290117016.
- Baruteau, A.-E.; Leurent, G.; Schleich, J.-M.; Gervais, R.; Daubert, J.-C.; Mabo, P. Can peripartum cardiomyopathy be familial? Int J Cardiol 2009, 137, 181–185, doi:10.1016/j.ijcard.2008.05.033.
- Haghikia, A.; Podewski, E.; Libhaber, E.; Labidi, S.; Fischer, D.; Roentgen, P. Phenotyping and outcome on contemporary management in a German cohort of patients with peripartum cardiomyopathy. Basic Res. Cardiol. 2013, 108, 366, doi:10.1007/s00395-013-0366-9.
- Morales, A.; Painter, T.; Li, R.; Siegfried, J.D.; Li, D.; Norton, N. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 2010, 121, 2176–2182, doi:10.1161/CIRCULATIONAHA.109.931220.
- Ntusi, N.A.; Wonkam, A.; Shaboodien, G.; Badri, M.; Mayosi, B.M. Frequency and clinical genetics of familial dilated cardiomyopathy in Cape Town: Implications for the evaluation of patients with unexplained cardiomyopathy. S Afr. Med. J. 2011, 1010, 394–398.
- Pearl, W. Familial occurrence of peripartum cardiomyopathy. Heart J. 1995, 129, 421–492, doi:10.1016/0002-8703(95)90032-2.
- Rajapreyar, I.; Sinkey, R.; Pamboukian, S.V.; Tita, A. Did a shared thioredoxin-reductase gene mutation lead to maternal peripartum cardiomyopathy and fatal dilated cardiomyopathy in her son? A case report. Case Rep. Womens Health 2020, 26, e00196, doi:10.1016/j.crwh.2020.e00196.
- van Spaendonck-Zwarts, K.Y.; van Tintelen, J.P.; van Veldhuisen, D.J.; van der Werf, R.; Jongbloed, J.D.; Paulus, W.J. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 2010, 121, 2169–2175, doi:10.1161/CIRCULATIONAHA.109.929646.
- Davies, J.E.; Winokur, T.S.; Aaron, M.F.; Benza, R.L.; Foley, B.A.; Holman, W.L. Cardiomyopathy in a carrier of Duchenne's muscular dystrophy. Heart Lung Transpl. 2001, 20, 781–784, doi:10.1016/s1053-2498(00)00240-0.
- Ahmed, A.; Spinty, S.; Murday, V.; Longman, C.; Khand, A. A de-novo deletion of dystrophin provoking severe 'peri-partum cardiomyopathy: The importance of genetic testing in peripartum cardiomyopathy to uncover female carriers. J. Cardiol. 2016, 203, 1084–1085, doi:10.1016/j.ijcard.2015.10.239.
- Cheng, V.E.; Prior, D.L. Peripartum cardiomyopathy in a previously asymptomatic carrier of Duchenne muscular dystrophy. Heart Lung Circ 2013, 22, 677–681, doi:10.1016/j.hlc.2012.11.015.
- Gurka, J.; Piherova, L.; Majer, F.; Chaloupka, A.; Zakova, D.; Pelak, O. Danon disease is an underdiagnosed cause of advanced heart failure in young female patients: A LAMP2 flow cytometric study. ESC Heart Fail 2020, doi:10.1002/ehf2.12823.
- Rowland, T.J.; Sweet, M.E.; Mestroni, L.; Taylor, M.R. Danon disease-dysregulation of autophagy in a multisystem disorder with cardiomyopathy. Cell Sci. 2016, 129, 2135–2143, doi:10.1242/jcs.184770.
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. Mol. Cell Cardiol. 2013, 58, 217–224, doi:10.1016/j.yjmcc.2012.12.009.
- Lim, K.R.Q.; Sheri, N.; Nguyen, Q.; Yokota, T. Cardiac Involvement in Dystrophin-Deficient Females: Current Understanding and Implications for the Treatment of Dystrophinopathies. Genes (Basel) 2020, 11, doi:10.3390/genes11070765.
- Moller, D.V.; Andersen, P.S.; Hedley, P.; Ersboll, M.K.; Bundgaard, H.; Moolman-Smook, J. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. J. Hum. Genet. 2009, 17, 1241–1249, doi:10.1038/ejhg.2009.34.
- Herman, D.S.; Lam, L.; Taylor, M.R.; Wang, L.; Teekakirikul, P.; Christodoulou, D. Truncations of titin causing dilated cardiomyopathy. N Engl. J. Med. 2012, 366, 619–628, doi:10.1056/NEJMoa1110186.
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Med. 2017, 19, 192–203, doi:10.1038/gim.2016.90.
- Tharp, C.A.; Haywood, M.E.; Sbaizero, O.; Taylor, M.R.G.; Mestroni, L. The giant protein Titin's role in cardiomyopathy: Genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Physiol. 2019, 10, 1436, doi:10.3389/fphys.2019.01436.
- Villard, E.; Perret, C.; Gary, F.; Proust, C.; Dilanian, G.; Hengstenberg, C. A genome-wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. Heart J. 2011, 32, 1065–1076, doi:10.1093/eurheartj/ehr105.
- Fang, X.; Bogomolovas, J.; Wu, T.; Zhang, W.; Liu, C.; Veevers, J. Loss-of-function mutations in co-chaperone BAG3 destabilize small HSPs and cause cardiomyopathy. Clin. Investig. 2017, 127, 3189–3200, doi:10.1172/JCI94310.
- Myers, V.D.; Tomar, D.; Madesh, M.; Wang, J.; Song, J.; Zhang, X.-Q. Haplo-insufficiency of Bcl2-associated Athanogene 3 in mice results in progressive left ventricular dysfunction, β-adrenergic insensitivity and increased apoptosis. Cell Physiol. 2018, 233, 6319–6326, doi:10.1002/jcp.26482.
- van Spaendonck-Zwarts, K.Y.; Posafalvi, A.; van den Berg, M.P.; Hilfiker-Kleiner, D.; Bollen, I.A.; Sliwa, K. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Heart. J. 2014, 35, 2165–2173, doi:10.1093/eurheartj/ehu050.
- Ware, J.S.; Li, J.; Mazaika, E.; Yasso, C.M.; DeSouza, T.; Cappola, T.P. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl. J. Med. 2016, 374, 233–241, doi:10.1056/NEJMoa1505517.
- Minoche, A.E.; Horvat, C.; Johnson, R.; Gayevskiy, V.; Morton, S.U.; Drew, A.P. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Med. 2019, 21, 650–662, doi:10.1038/s41436-018-0084-7.
- Kryczka, K.E.; Dzielińska, Z.; Franaszczyk, M.; Wojtkowska, I.; Henzel, J.; Śpiewak, M. Severe course of peripartum cardiomyopathy and subsequent recovery in a patient with a novel TTN gene-truncating mutation. J. Case Rep. 2018, 19, 820–824, doi:10.12659/AJCR.909601.
- Nishimoto, O.; Matsuda, M.; Nakamoto, K.; Nishiyama, H.; Kuraoka, K.; Taniyama, K. Peripartum cardiomyopathy presenting with syncope due to Torsades de pointes: A case of long QT syndrome with a novel KCNH2 mutation. Med. 2012, 51, 461–464, doi:10.2169/internalmedicine.51.5943.
- Kim, J.; Reutrakul, S.; Davis, D.B.; Kaplan, E.L.; Refetoff, S. Multiple endocrine neoplasia 2A syndrome presenting as peripartum cardiomyopathy due to catecholamine excess. J. Endocrinol. 2004, 151, 771–777, doi:10.1530/eje.0.1510771.
- Glocklhofer, C.R.; Steinfurt, J.; Franke, G.; Hoppmann, A.; Glantschnig, T.; Perez-Feliz, S. A novel LMNA nonsense mutation causes two distinct phenotypes of cardiomyopathy with high risk of sudden cardiac death in a large five-generation family. Europace 2018, 20, 2003–2013, doi:10.1093/europace/euy127.
- Molina, P.; Sanz-Sanchez, J.; Fenollosa, M.; Martinez-Matilla, M.; Giner, J.; Zorio, E. Arrhythmogenic cardiomyopathy with left ventricular involvement versus ischemic heart disease: Lessons learned from the family study and the reviewed autopsy of a young male. Forensic Sci. Res. 2019, 4, 274–279, doi:10.1080/20961790.2019.1616247.
- Murakami, T.; Hayashi, Y.K.; Noguchi, S.; Ogawa, M.; Nonaka, I.; Tanabe, Y. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Neurol. 2006, 60, 597–602, doi:10.1002/ana.20973.
- Robyns, T.; Willems, R.; Van Cleemput, J.; Jhangiani, S.; Muzny, D.; Gibbs, R. Whole exome sequencing in a large pedigree with DCM identifies a novel mutation in RBM20. Acta Cardiol. 2019, 1–6, doi:10.1080/00015385.2019.1674490.
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Rev. Cardiol. 2020, 17, 286–297, doi:10.1038/s41569-019-0284-0.
- Paldino, A.; De Angelis, G.; Merlo, M.; Gigli, M.; Dal Ferro, M.; Severini, G.M. Genetics of dilated cardiomyopathy: Clinical implications. Cardiol. Rep. 2018, 20, 83, doi:10.1007/s11886-018-1030-7.
This entry is adapted from the peer-reviewed paper 10.3390/genes12010103