2. Implication of HCs and Panx1 Channels in Disturbances of Ionic and Redox Homeostasis as Well as Pro-Inflammatory and Pro-Fibrotic Signaling
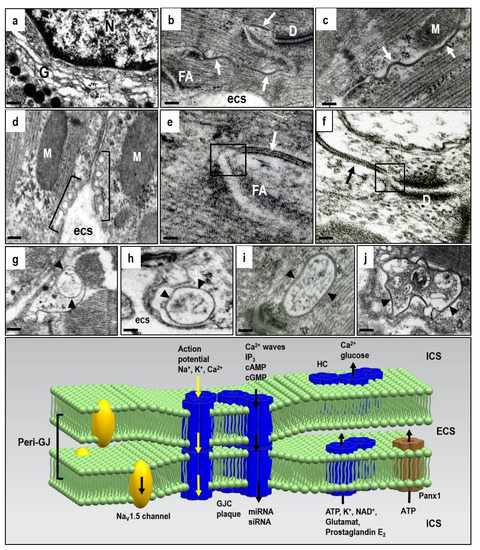
Compared to GJCs, the HCs and Panx1 channels operate out of cardiomyocyte membrane gap junctions. Therefore, dominant myocardial HCs and Panx1 channel topology appears restricted to the lipid rafts/caveolae on lateral cardiomyocyte plasma membrane (d). In addition, they also act at the peri-nexus [
22], i.e., at the peri-gap junctional membrane (e,f). HCs also perform the “non-canonical” functions of Cx43, beyond those of the GJCs and independent of gap-junction formation [
25,
55,
56].
Findings from various non-cardiac tissues that might be relevant for cardiomyocytes indicate that HCs channels remain closed in physiological resting conditions, but change to open conformation as the extracellular Ca
2+ concentration decreases or intracellular Ca
2+ concentration increases, which open Panx1 channels as well [
57,
58,
59,
60,
61] However, HCs and Panx1 channels do not have the exact same regulation. HCs tend to be activated by strong depolarization, while Panx1 channels activity may be induced at the resting membrane potential [
62]. The pore diameter increases from 1.8 to 2.5 nm and this enables transmission of molecules up to 1 kD. HCs opening enables typical single-channel conductance of approximately 220 pS [
63] and Panx1 channels have a large unitary conductance of around 500 pS. In addition, hydrophobic extracellular domains are also crucial in regulating Ca
2+-dependent conformational changes [
20].
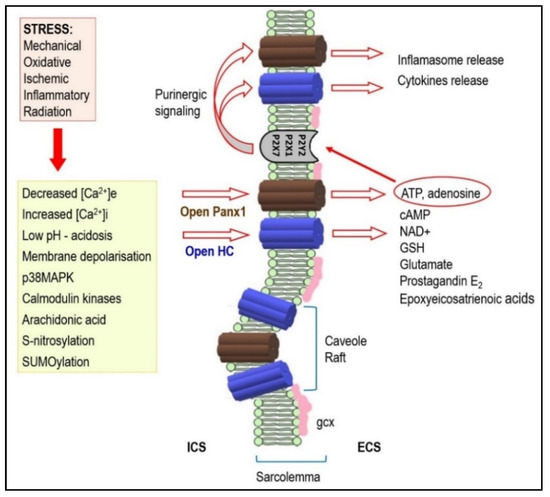
The HCs and Panx1 channels can open following different extracellular stimuli. This is established in various non-cardiac tissues [
57,
58,
59,
64,
65], but highly relevant to the heart. The HCs and Panx1 channels can therefore be activated by membrane depolarization, metabolic inhibition, and stresses including shear mechanical membrane forces and ionic and ischemic stress [
24,
38,
57,
62,
66,
67]. The HCs, and most likely also the Panx1 channels, are influenced by intra- and extra-cellular pH and phosphorylation and redox status [
22,
24,
37,
58,
66,
68,
69,
70]. All these events during heart disease development can activate both HCs and Panx1 channels and promote arrhythmias through bi-directional ion passage and small metabolic or signaling molecules below 1–2 kDa. Thus, HCs and/or Panx1 channels can mediate Na
+, K
+, Ca
2+, cAMP, ATP, NAD+ influx as well as the transmission of glutamate, glutathione, prostaglandin-E2 and epoxyeicosatrienoic acid [
24,
35,
71]. Panx-1 channels are known mostly as ATP release channels [
60]. Intracellular localized Panx1 has also been proposed as a regulator of sarcoplasmic reticulum-based Ca
2+ homeostasis [
72].
HCs and Panx1 channel opening can profoundly affect intracellular ionic homeostasis, contribute to Na
+ and Ca
2+ loading and K
+ loss, alter redox status, and facilitate pro-inflammatory and pro-fibrotic conditions [
54,
57,
58,
62,
73]. These events impair GJCs function and produce myocardial electrical instability, thereby promoting malignant arrhythmias [
4,
32,
66,
74,
75]. It was also previously noted that the activation of peri-junctional Panx1 channels [
37] and Na
v 1.5 channels can influence cardiac conduction [
36,
39]. Abnormal Na
v1.5 channel function along with autoantibodies against Cx43 and activation of Panx1 channels may underlie heritable arrhythmic syndromes [
76,
77,
78].
HCs and Panx1 channel mediated ATP release is fundamental in the purinergic signaling which is so important in promoting inflammation in vascular function/dysfunction [
62,
65,
66,
79,
80,
81]. The inflammation is known to impair GJCs function and significantly contribute to arrhythmogenesis [
74]. The Panx1 channels present at internal membranes may be implicated in K
+ influx to mitochondria and intracellular Ca
2+ leak/release from the endoplasmic reticulum [
35,
71,
82]. Moreover, ATP binding to the P2Y receptors increases inositol 1,4,5-triphosphate which then releases Ca
2+ from the ER and enhances ATP signaling to neighboring cells [
24]. Disturbances in intracellular ionic homeostasis and Ca
2+ overload promote triggered “post-depolarization” and GJCs uncoupling which render the heart prone to malignant arrhythmia development; and especially VF [
83,
84,
85,
86,
87]. This triggered Cx activity and GJCs disordered expression and dysfunction are also implicated in AF development [
85,
88].
Cellular stress, including myocardial ischemia, enhances cardiomyocyte ATP release through the Panx1 channels that facilitate early fibroblast activation [
66]. This suggests an early paracrine event leading to pro-fibrotic responses which could be involved in arrhythmogenic substrate development. In contrast, HCs inhibition by TAT-Gap19 alleviated tissue fibrosis and introduced the possibility of preventing arrhythmias [
89]. Fibrosis precedes inflammation, and cardiac fibroblasts contribute to the inflammatory milieu through increased secretion of pro-inflammatory cytokines and chemokines released by the HCs and Panx1 channels [
90,
91]. Here, the Panx1 channels are interesting because of their implication in a variety of cellular responses [
24,
38], including apoptosis [
92], inflammation [
93], and innate immune processes [
94,
95]. Inflammation is also a prominent feature of arrhythmogenic cardiomyopathy [
96]. Therefore, targeting inflammatory pathways could be an effective new mechanism-based therapy for familial non-ischemic heart muscle disease which causes sudden death in the young, and especially in athletes.
illustrates that functional HCs and Panx1 channels provide paracrine and autocrine communication pathways for ion and small molecule passage across the plasma membrane [
22,
37,
38,
66,
97]. HCs and Panx1 channels may regulate many cellular processes but little information on their mechanisms is available. Extended information on HCs and Panx1 channel function from in vitro sources indicates their implication in diverse physiological and pathological responses relevant to the cardiovascular system [
20,
22]. However, the cardiac muscle remains ‘terra incognita’ and this provides challenges. This is especially important, in conjunction with rafts/caveolae related signal-transducing molecules, in the development of heart disease and life-threatening arrhythmias. The compartmentalized signal transduction appears to be an attractive area of research.
Figure 2. Under stressful conditions, several intracellular signals regulate hemi-channels (HCs) opening. Activated HCs enables release of signaling molecules to the extracellular environment. Therefore, it acts as a paracrine and autocrine communication pathway. HCs mediated ATP release is important in the purinergic signaling which is so important in cardiac pathophysiology. It may also be implicated in arrhythmogenesis (see text for details). Abbreviations: ICS—intracellular space, ECS—extracellular space, gcx—glycocalyx.
It is important that the caveolae and lipid rafts of cholesterol and sphingomyelin enriched membrane micro-domains are considered in HCs and Panx1 channels. These are all involved in the regulation of cell signaling pathways [
58,
59,
98,
99] and in GJCs homeostasis [
100,
101,
102,
103,
104]. Caveolin-3 is the major caveolin isoform in cardiomyocytes, and this has been implicated in 17β-estradiol-elicited, rapid signaling to regulate Cx43 phosphorylation during ischemia [
101]. Moreover, the caveolae/lipid rafts have been involved in reactive oxygen species production, redox signaling and K
+, K
ATP, Na
+, and Ca
2+ ion channel functions [
105,
106,
107].
Cardiomyocytes and endothelial capillary cells are rich in caveolae which invaginate the plasma membrane, and this suggests the implication of these compartments in extensive HCs and Panx1 channel-mediated paracrine and autocrine signaling. The interaction of these channels and caveolins implies compartmentalized signaling, and this could be most important in the pathophysiological development of heart dysfunction and arrhythmias [
45,
103,
104,
108,
109,
110]. Investigation of this issue presents future challenges.
3.Impact ofHCs and Pannex1 Channels Activity onDevelopment of Cardiac Arrhythmias
There is still a lack of information on HCs and Panx1 channel roles in cardiac muscle.
Nevertheless, it has been shown that both HCs and Panx1 channels are the potential
route for Na+ inflsux and K+ efflux during ischemia in isolated ventricular cardiomyocytes [39,40,62,128]. This can induce electrical disturbances [14,41]. The opening of HCs and Panx1 channels during ischemia has also contributed to re-perfusion injury following brief cardiomyocyte ischemia [66,91,129–131]. The activation of HCs and Pannex1 channels during ischemia/hypoxia, combined with other metabolic inhibition and post-ischemic reperfusion mechanisms may compromise cardiomyocyte ability tomaintain ionic homeostasis. This is an essential step in promoting both arrhythmias and apoptosis [87,91,131–134].
Metabolic inhibition followed by pro-arrhythmogenic [Ca2+]i and [Na+]i overload in isolated cardiomyocytes was significantly reduced by halothane which decreases HCsconductance [135]. Importantly, HCs contribute to cytoplasmic Ca2+ oscillations by providinga bimodal Ca2+ dependent Ca2+ entry pathway [136]. Moreover, ischemia and/or hypoxia are associated with increased production of NO and S-nitrosylation [137,138] which is important in regulating HCs and Panx1 channel permeability [139,140]. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential [141].
The NADPH oxidase inhibitor apocynin prevented HCs activity by reducing nitroso-redox stress [142]. All these factors strongly suggest that HCs are implicated in arrhythmogenesis.
The release of ATP via HCs or Panx1 channels in pathological conditions could induce post-depolarization triggered activity. This would generate ventricular tachycardia or VF [145,146]. Moreover, purinergic receptor activation by ATP induces ventricular tachycardia through membrane depolarization and Ca2+ homeostatic disorders [147]. Resultant sporadic Panx1 channel opening then triggered action potentials and promoted arrhythmogenic activity [23].
In addition, extracellular ATP induced shortened action potential duration in tissue preparations of atrial and ventricular myocardium and in the myocardial sleeves of pulmonary veins [148]. These events are known to precede AF [85,117]. The implication of ATP or adenosine in AF development, and its post-ablation recurrence, have been
reported [149–153]. This AF was associated with a strong increase in atrial adenosine [154].
In addition, non-myocytes in scar tissue can be electrically coupled to cardiomyocytes
through GJCs, HCs, and Panx1 channels and contribute to electrotonic conduction
across scar tissue [158,159]. Cx43 is increased in activated fibroblasts during fibrogenesis
[91,160,161] and it enhances the possibility of electrically coupled cardiomyocytes and
fibroblasts [160–162]. This hetero-cellular electrotonic coupling may facilitate conduction
disturbances and arrhythmias [163]. Fibroblasts and myofibroblasts can alter cardiomyocyte
excitation-contraction coupling through paracrine mechanisms [164], and this suggests
the paracrine modulation of myocardial contractility and synchronized contraction.
Fibrotic tissue, however, is an inefficient electrical signal conductor and cannot rhythmicallycontract, thereby jeopardizing critical myocardial functions [91]. This combination provides
heterogeneous cardiac tissue, and this is a well-known predictor of arrhythmia risk.Although HCs and
Panx1 channels present an attractive therapeutic target, their inhibition requires the development
of more specific inhibitory agents.