The diarylurea is a scaffold of great importance in medicinal chemistry as it is present in numerous heterocyclic compounds with antithrombotic, antimalarial, antibacterial, and anti-inflammatory properties.
- diarylureas
- antitumor agents
1. Introduction
Ureas (R-NHCONH-R’) are known organic compounds that possess biological activities and serve as templates for numerous medicinal chemistry researches [1]. Barbital is a diethylmalonyl urea discovered at the beginning of 1900, used as sleep aid and hypnotic [2]. In the following century, the urea scaffold has represented the pharmacophore the backbone motif for entire classes of therapeutic agents [3].
Diarylureas are found in numerous heterocyclic compounds with various biological activities [4], such as antithrombotic, antimalarial, antibacterial, antinflammatory, and anticancer [5,6]. In particular, diarylurea is a prominent pharmacophore in anticancer drugs. This activity is due to its near-perfect binding with certain acceptors. The NH moiety behaves as hydrogen bond donor and the urea oxygen atom acts as acceptor (Figure 1) [7].
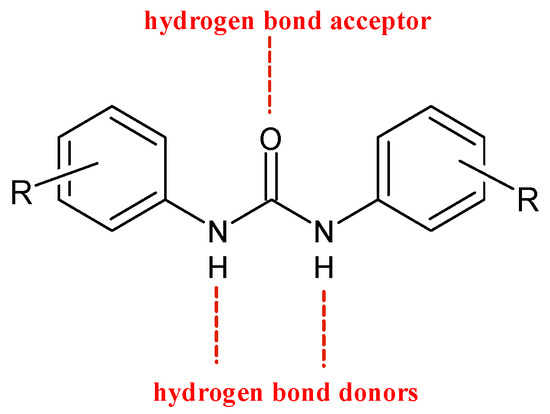
Figure 1. H-bonds in diarylureas.
This structure provides urea derivatives endowed with capability of binding several enzymes and receptors [8,9,10]. Moreover, it may link different pharmacophore fragments of new biological active compounds. A urea linker has been used to overcome the poor solubility of some phenyl N-mustards [11]. In this way, the authors obtained water soluble N-mustards, some of which showing high anticancer activity against various human tumor xenograft models and were able to introduce cross-linking within the DNA double strand. Diarylurea-based compounds present strong inhibitory activity against kinases, including RAF kinases [12], platelet derived growth factor receptor (PDGF) [13], vascular endothelial growth factor receptor 2 (VEGFR-2) [14], receptor tyrosine kinase (RTKs) [15], and Aurora kinases [16]. The diarylurea moiety is, in fact, widespread in type II kinase inhibitors. These compounds circumvent kinases in an inactive state, the so-called DFG-out, and occupy a hydrophobic pocket next to the ATP-binding site. The diarylurea fragment is able to link the hinge-binding moiety with the portion that occupies the hydrophobic pocket that is in the inactive conformation of kinases [17] (Figure 2).
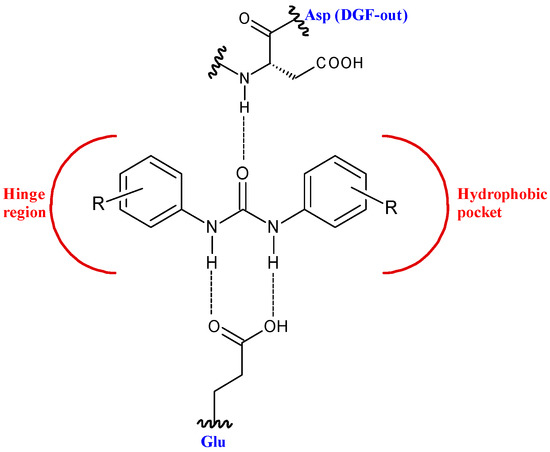
Figure 2. Diarylureas in the type II kinase inhibitor.
Diarylureas represent the skeleton of the main systemic therapies for several cancers, as advanced, metastatic hepatocellular carcinoma (HCC) [18], advanced renal cell carcinoma (RCC) [19], gastrointestinal stromal tumors (GISTs) [20], metastatic colorectal cancer (mCRC) [21]. Sorafenib is a multi-targeted small molecule tyrosine protein kinase that improves median survival over placebo for unresectable HCC patients [22]. In 2008 it obtained Food and Drug Administration (FDA) and European Medicinal Agency (EMEA) approval for the treatment of RCC and HCC [23]. Basing on sorafenib as the lead compound, several other diarylurea derivatives, such as regorafenib, linifanib, tivozanib, and ripretinib have been synthesized and evaluated as kinase inhibitors. Regorafenib was approved by the FDA in the United States in February 2013 in patients with advanced GISTs for those who had failed on imatinib and sunitinib [24,25]. Linifanib is currently being studied in HCC clinical trials [26]. Unlike the other inhibitors of VEGFR and PDGFR, linifanib seems to be also involved in adipocyte browning, thus being considered for the treatment of obesity [27]. Tivozanib is a diarylurea which is to be considered as third and fourth line therapy in patients with metastatic RCC in phase 3 study [28]. By end of 2019, the Chinese and European regulatory authorities have marketed five small-molecule protein kinase inhibitors (PKIs), including tivozanib [29]. Ripretinib has been suggested as a promising treatment for advanced GISTs [30].
2. Diarylureas in Therapy or in Clinical Studies as Anticancer Agents
2.1. Sorafenib
Sorafenib (BAY-43-9006, Nexavar®, Figure 3) is an oral receptor TKI that determines the inhibition of Raf serine/threonine kinases and receptor tyrosine kinases (VEGF 1, 2, 3 and PDGF-β, FMS-like tyrosine kinase-3 (FLT-3), and c-KIT) that are components of signaling pathways controlling tumor growth and angiogenesis [31].
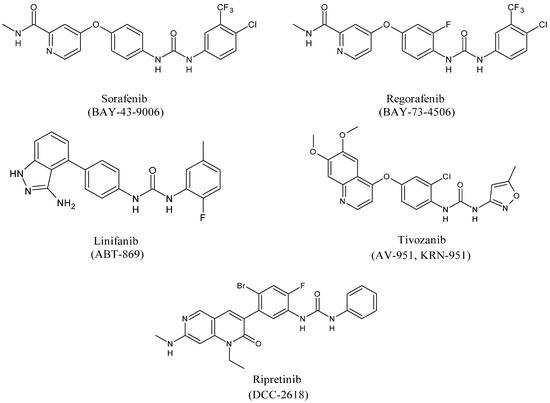
Figure 3. Structures of diarylureas.
Sorafenib inhibits the kinase activity of C-RAF and B-RAF (wild type and V600E mutant) showing IC50 = 6.22 and 38 nM, respectively. This compound is considered the most important drug in the late stage of injury for advanced stages of HCC [32] which is the second cause of cancer-related mortality all over the world [33]. For more than ten years, sorafenib has been the sole systemic treatment for advanced HCC [34]. However, some advanced HCC patients do not respond to therapy with sorafenib. Thus, combination studies of sorafenib with other drugs have been studied. Combined treatment with interferon-lambda 3 (IFN-λ3) and sorafenib show an effect of synergism in suppressing HCC cancer growth and in the promotion of cell apoptosis in vitro and in vivo [35]. A more recent study demonstrated that combination immunotherapy of sorafenib with atezolizumab, an immune checkpoint inhibitor (ICI) that target the programmed-cell death-1 receptor/ligand (PD-1/PD-L1) pathway, and bevacizumab, an anti-VEGF mAb, is superior to sorafenib alone as the first-line therapy of advanced HCC [36]. Moreover, given that liver function is essential for a correct prognosis, a precise rating for the safe prescription and clinical development of ICI in HCC is required. Recently, the albumin-bilirubin (ALBI) grade was used as an alternative biomarker for the prognosis [37]. The efficacy of sorafenib is limited by several factors as systematic tolerance and the poor solubility in water. Furthermore, the hydrophobicity of sorafenib is responsible of its low bioavailability as it decreases the absorption by the gastrointestinal tract. Nexavar® (Bayer Healthcare Pharmaceuticals–Onyx Pharmaceuticals) is used as tablets containing sorafenib tosylate to slightly improve the solubility. In order to increase the solubility and bioavailability of the drug, sorafenib-loaded lipid-based nanosuspensions were used [38]. The administration of sorafenib to the target cells could ameliorate patient survival and reduce the further proliferation of the tumor [39]. Thus, a drug delivery system for sorafenib has been recently studied in order to help the administration of therapies in malignant cells and raise its clinical efficacy [40]. Recently, the treatment of cancers of the gastrointestinal tract during the COVID-19 pandemic [41] has been studied: coronavirus-adapted institutional recommendations have been formulated. Sorafenib was recommended in the first-line setting only in patients with disease subtype Eastern Cooperative Oncology Group performance status (ECOG PS) 0 or 1 and Child-Pughscore A hepatocellular carcinoma [42]. In spite of its selectivity, sorafenib can determine adverse effects, such as severe respiratory and liver failure, fatigue, stomatitis, hand-foot syndrome, diarrhea, and myelosuppression, thus posing a challenge for oncologists [43]. The therapy with sorafenib might be done ad hoc to increase the therapeutic effects and reducing adverse effects [44]. Sorafenib is also studied for iodine-resistant advanced thyroid carcinoma [45]. Finally, it is important to note that sorafenib is also involved in cytoskeleton alteration that leads to cancer cells death by apoptosis. Wang et al. [46] reported that the treatment of Hep3B and PLC/PRF/5 human hepatoma cells with sorafenib induces a drastic loss of actin fibers and the redistribution of F-actin around the cell nuclei. This effect is due to the regulation of protein kinases and phosphatases that ends with cofilin dephosphorylation, which is an actin-binding factor necessary for the reorganization of actin. Chen et al. [47] demonstrated that the ability of sorafenib in inducing human prostate cancer cell line PC-3 apoptosis, through cytoskeleton destabilization, increased if combined with zinc exposure, suggesting that zinc may sensitize prostate cancer cells to sorafenib treatment. D’Alessandro et al. [48] demonstrated a synergistic effect on HCC cells migration using combined doses of sorafenib and/or vitamin K1 with insulin like growth factor I receptor (IGF1-R) antagonists enhancing the reduction and reorganization of F-actin, probably through the modulation of MAPK cascade.
2.2. Regorafenib
Regorafenib (BAY 73-4506, Stivarga®, Figure 3) is the fluorinated analogue of sorafenib. It is an orally active diphenylurea multikinase inhibitor that targets stromal (PDGFR-β, FGFR-1), angiogenic (VEGFR1-3, TIE-2), and oncogenic receptor tyrosine kinases (c-KIT, RET, and RAF-1) [49]. It is the first multi-targeting kinase inhibitor which was approved by FDA in 2012 for the treatment of mCRC patients in refractory to standard chemotherapy [50]. In addition, regorafenib treatment determined an important amelioration in progression-free survival (PFS) in comparison with placebo in patients with metastatic GISTs after standard treatments; thus, it has also received FDA-approval for this indication since 2013. Then, in 2017, FDA approved regorafenib as a therapy for patients with advanced HCC [51]. Regorafenib showed a significant amelioration of PFS and overall survival (OS) in comparison with placebo. Regorafenib has been described in phase II clinical trials in different tumors, including RCC, soft-tissue sarcoma (STS), second- and third-line treatments for medullary thyroid cancer. However, several post-marketing observational studies, after the treatment of mCRC patients showed extensive data of toxicities (CORRELATE, REBECCA, RECORA, Japanese post-marketing study) [52,53,54]. It was found that adverse reactions due to regorafenib frequently occurred in the initial stages of treatment, mostly in the first cycle [55]. Thus, in patients with mCRC during the first cycle of regorafenib, the use of a dose-escalation strategy treatment was suggested [56]. This strategy was then supported by a multicenter, open-label, phase II study [57]. Recently, during the COVID-19 outbreak, regorafenib has been considered as a therapy for gastrointestinal cancer [42]. A drug-delivery system has been studied for regorafenib, too [40]. The pivotal RESORCE (NCT01774344) phase III trial studied regorafenib therapy in patients with HCC who were tolerant to sorafenib, but who had progressed during sorafenib treatment [58]. Regorafenib has been demonstrated to inhibit glioblastoma multiforme (GBM) growth through PSAT1-mediated autophagy arrest [59], and to have beneficial effect in Alzheimer’s disease (AD) and formation of dendritic spine in vitro and in vivo [60].
2.3. Linifanib
Linifanib (ABT-869, Abbott Laboratories, Abbott Park, IL, USA, Figure 3) is an orally available TKI which targets VEGFR and PDGFR with relevant specificity and low off-target inhibition. Linifanib can also inhibit FLT-3 [61]. It does not show significant activity against representative cytosolic tyrosine and serine/threonine kinases [62]. Linifanib is a colony-stimulating factor-1 receptor (CSF-1R) inhibitor through the inhibition of the phosphorylation of CSF-1R tyrosine kinase in transfected cells [63]. It is used as a therapy for non-small cell lung carcinoma (NSCLC), liver cancer, breast cancer, colorectal cancer [45]. Preclinical and early clinical trials showed interesting activity in various human neoplasms with a satisfactory profile of toxicity. Linifanib competes with ATP in the binding site domain of tyrosine kinase, thus it prevents downstream signaling [64]. Phase II trial studies show that linifanib is useful for the treatment of patients with advanced, refractory colorectal cancer that expresses k-Ras mutations [65]. In an open-label phase II trial linifanib showed interesting clinical activity, as monotherapy, in patients with advanced HCC [66]. Linifanib versus sorafenib was studied in terms of efficacy and tolerability. Linifanib and sorafenib showed similar OS in advanced HCC. Linifanib did not meet predefined superiority and non-inferiority OS boundaries; thus, the study did not reach the primary end point. Secondary end points, time to progression (TTP), and objective response rate (ORR), favored Linifanib; safety results favored sorafenib [67]. Although linifanib is currently examined in HCC clinical trials, it has not yet been studied in preclinical and clinical studies for gastric cancer. 5-Fluorouracil (5-FU) and cisplatin represent the first-line chemotherapy for patients with gastric cancer and the combined use with linifanib inhibits synergistically the viability of some gastric cancer cell lines and led to remarkable suppression of VEGF-induced angiogenesis in vitro and in vivo [68]. Linifanib has demonstrated to be also useful in the treatment of anaplastic thyroid cancer (ATC), that is considered the most aggressive form of thyroid cancer. The synergistic use of linifanib and irinotecan significantly increased the survival of ATC-affected mice. These observations have been made by using an orthotopic in vivo model that better recapitulates features of human tumors than the more simplistic subcutaneous xenograft models, suggesting a potential role of this co-treatment in ATC patient’s treatment [69]. Finally, linifanib was demonstrated to interfere with adipocyte browning. It suppresses STAT3 signaling pathway, thus leading to the enhancement of adipocyte browning and inhibition of adipogenesis. Linifanib’s blocking browning effect was demonstrated as the phosphorylation of STAT3 was reduced by linifanib and the STAT3 activator SD19, as well [27].
2.4. Tivozanib
Tivozanib (AV-951, KRN-951, FOTIVDA®, Figure 3), used as the hydrochloride monohydrate salt, is a bioavailable inhibitor of angiogenesis which targets VEGFR tyrosine kinases with high antitumor activity. It is a VEGF-TKI specific for VEGFR1–3, showing an inhibitor effect at nanomolar concentrations, with IC50 values of 30 nM, 6.5 nM, and 15 nM for VEGFR1, 2 and 3, respectively. The compound is unique in that it is highly specific for VEGFR1–3, and presents minimal residual effects on c-KIT and PDGFR-β [70]. It presents a long half-life, too [71]. It has shown considerable efficacy for the treatment of advanced RCC over the past decade. In August 2017, tivozanib was approved by the EMEA as a first-line therapy for patients with advanced RCC and those who are VEGFR and mTOR pathway inhibitor-naïve following disease progression after previous therapy with cytokines for advanced RCC. Tivozanib was compared with sorafenib in a phase III trial for patients with metastatic RCC. Tivozanib improved PFS, but not OS, and showed a differentiated safety profile, in comparison with sorafenib, as initial targeted therapy for metastatic RCC [72]. Preclinical data and phase III trials of tivozanib in RCC, TIVO-1, and TIVO-3 have been recently summarized. Given the agent’s excellent tolerability profile it is appropriate for those patients with heavily pretreated disease that could exhibit clinical deterioration. Currently, the standard therapy is represented by nivolumab and ipilimumab, followed by cabozantinib. Tivozanib may represent a third-line treatment after failure of these agents [73]. The results of phase III TIVO-3 trial (American Society of Clinical Oncology Virtual Scientific Program, 2020) showed that tivozanib significantly improved PFS, compared with sorafenib, in patients with highly relapsed or refractory metastatic RCC [74,75,76,77]. Tivozanib activity has been also investigated in hepatocellular carcinoma in association with durvalumab [78] and in recurrent, platinum-resistant ovarian cancer, fallopian tube cancer, and primary peritoneal cancer [79,80]. Tivozanib has been studied in phase I and II clinical trials as monotherapy and in combination with other drugs for the treatment of STS [81], glioblastoma [82], breast [83], and colorectal cancers, and other advanced gastrointestinal cancers [84,85].
2.5. Ripretinib
Ripretinib (DCC-2618, QINLOCKTM, Figure 3) is an oral inhibitor of tyrosine kinase that primarily inhibits KIT proto-oncogene receptor tyrosine kinase and platelet-derived growth factor receptor A (PDGFRA) kinase signaling. Ripretinib also inhibits other kinases, such as PDGFRB, TIE2, VEGFR2, and BRAF. It was designed for cancers and myeloproliferative neoplasms, especially GISTs. Ripretinib is a “switch-control” kinase inhibitor that forces the activation loop (or activation “switch”) into the inactive conformation. In preclinical cancer models it has shown efficacy, and preliminary clinical data show that Ripretinib inhibits a broad range of KIT mutants in patients with drug-resistant GISTs [86]. The INVICTUS study demonstrated the efficacy and safety of ripretinib as the fourth-line treatment versus placebo in patients with advanced GISTs [87]. In May 2020, ripretinib received approval from the US FDA for the treatment of patients with advanced GISTs who had received previous treatment with more than two kinase inhibitors [88]. Ripretinib is being evaluated in an ongoing phase III study (INTRIGUE) as a second-line therapy in comparison with sunitinib after progressing on imatinib [89]. Recently, ripretinib is being investigated in clinical trials for systemic mastocytosis (SM) [90], and has been also proposed for the treatment of STS [91].
2.6. Mechanisms of Inhibition of Diarylureas
The proposed inhibitory mechanisms of diarylureas depend on their structure. Garuti et al. [5] reported the crystal structure of V600EB-RAF kinase domains in complex with sorafenib. The pyridyl ring is shown to occupy the ATP adenine-binding pocket and to interact with three amino acids residues. The trifluoromethyl phenyl, that is a lipophilic moiety, fits into a hydrophobic pocket. The urea moiety forms two hydrogen bonds with V600EB-RAF, one with the aspartate, and one with the glutamate residue. Recently, the 2D interaction of the co-crystallized sorafenib inside the active site of B-Raf has been reported [92]. Regorafenib differs from sorafenib only for a fluorine atom, thus its interactions are similar to those of sorafenib. Chen et al. (2017) proposed an alternative mechanism for colorectal cancer for regorafenib. It seems that it interacts with microRNA-21 (miR-21), an oncogenic miRNA which plays a crucial role in resisting programmed cell death in CRC cells. RNA–ligand docking, molecular dynamics simulation showed that regorafenib can directly bind to miR-21 pre-element [93]. Docking studies of linifanib with FLT3 were recently reported, evidencing that 3-amino-indazole interacts with the ATP-bind site [61]. Kajal et al. (2018) has reported the 2D co-crystal-binding conformation of VEGFR2-Tivozanib, in which tivozanib mimics the binding pattern of ATP [94]. Finally, studies on the mechanism of action of ripretinib have been recently reported [86].
This entry is adapted from the peer-reviewed paper 10.3390/app11010374