The blockade of immune checkpoints (ICPs), such as cytotoxic T lymphocyte associated protein-4 (CTLA-4) and programmed death-1 (PD-1) and its ligand (PD-L1), has propelled the field of immuno-oncology into its current era.
- LAG-3,
- immune checkpoint,immunotherapy
1. Introduction
Immune cells are controlled by a plethora of molecules that act as “security brakes” at multiple stages of the immune response. These regulations are important to prevent the destruction of tissues caused by inappropriate and/or disproportionate responses to invading pathogens. Cancer cells exploit such inhibitory immune checkpoints (ICPs) to escape destructive tumor-specific immune responses, which is unfavorable for the patients’ outcome.
In 2018, cancer caused 9.6 million deaths and was diagnosed in 18.1 million patients worldwide[1]. Cancer treatments focusing on reinvigorating exhausted tumor-specific immune cells significantly improved the survival of patients. In particular, the anti-tumor effects observed after monoclonal antibody (mAb)-mediated the blockade of the ICPs; cytotoxic T lymphocyte associated protein-4 (CTLA-4, CD152), programmed death-1 (PD-1, CD279) and its ligand PD-L1 (CD274, B7-H1), revolutionized the field of immuno-oncology. Therapeutic success was achieved in a fraction of patients and provided clinical evidence that corroborated the preclinical finding that ICPs exert inhibitory effects on immune cells that try to eradicate cancer cells. However, the response frequencies to these widely used ICP-blocking mAbs are suboptimal as a consequence of, amongst others, tumor resistance, an absence of tumor-infiltrating lymphocytes (TILs), and the presence of inhibitory myeloid cells[2]. Moreover, the occurrence of immune-related adverse events (irAEs) has been a reason to discontinue the use of ICP-blocking mAbs. Taken together, researchers are continuously untangling the biology of inhibitory receptors to increase response rates and to prevent irAEs.
The list of inhibitory ICPs that negatively regulate anti-tumor immune responses is growing. Among these next-generation ICPs, lymphocyte activating gene-3 (LAG-3, CD233) has emerged as an eminent target in the development of cancer treatment and holds substantial prognostic value. With as many as 15 different compounds targeting LAG-3 under (pre)clinical evaluation, it is the most widely studied ICP next to CTLA-4 and PD-1/PD-L1. LAG-3, also known as CD223, is expressed on numerous immune cell types, where its exact mechanism of action is yet to be fully discovered. However, it is certain that LAG-3 shows a remarkable synergy with PD-1 in promoting immune escape of cancer cells[3][4][5][6]. Notably, the simultaneous blockade of LAG-3 and PD-1 has shown striking clinical results in melanoma patients that were not responding well to initial PD-1 or PD-L1 monotherapy [7][8].
The growing list of inhibitory ICPs brings forth the challenge of defining which patients are likely to benefit from ICP therapy and which inhibitory ICP(s) prevail(s) in the patients’ tumors and therefore should be targeted. Presently, patients are selected for treatment with ICP-blocking mAbs, depending on the level of inhibitory ICP expression in the tumor microenvironment (TME), which is determined using immunohistochemistry (IHC). The static picture created by the IHC of a tumor biopsy does not take in account the heterogeneously distributed expression of ICPs. The latter can explain the fact that “non-expressors” do respond to ICP-blocking therapies[9]. Contrarily, some patients with IHC samples that show a clear expression of ICPs in the TME are observed not to react to ICP-blocking mAbs[10][11]. This can be due to amongst others, the compensatory expression of other ICPs[9][12][13]. Recent studies explored the field of molecular imaging as a more suited alternative for the non-invasive detection of ICPs in cancer patients. Molecular imaging allows the whole body visualization of prognostic or predictive markers. It can be performed non-invasively and repetitively, which allows quantifying and tracking of the dynamic evolution of expression patterns.
The aim of this review is to provide an overview of the biology of LAG-3 in the context of cancer as well as to provide an overview on the strategies used to detect and block LAG-3.
2. The Receptor LAG-3 and Its Interaction Partners
LAG-3, also called CD223, was identified in 1990 as a receptor that was expressed on a natural killer (NK) cell line cultured with interleukin (IL)-2[14]. A close relationship with the cell surface protein CD4 was shown in terms of location and organization of its coding region as well as in terms of amino acid sequence and protein structure. The LAG-3 gene is located on the distal portion of the short arm of human chromosome 12 (12p13.31), which is adjacent to the coding region for CD4, and contains eight exons[15]. The conservation of LAG-3 among species is shown by its 70% and 78% homology with murine[14] and pig[16] LAG-3, respectively. The protein LAG-3 is 503 amino acids large, weighs 70 kDa, and is a type I transmembrane protein that contains four extracellular Immunoglobulin (Ig)-like domains termed domain 1 to 4. Approximately 20% of its amino acid sequence is identical to the CD4 protein, which is mostly pronounced in the extracellular region[14][17]. In contrast, the intracellular region of CD4 and LAG-3 lack homology, suggesting different functions. Due to the extracellular similarity, CD4 and LAG-3 share the same ligand, i.e., major histocompatibility complex class II (MHC-II) proteins. In LAG-3, a 30 amino acid long “extra loop” in domain 1 has been reported to engage MHC-II[18][19]. Notably, LAG-3 does not universally recognize MHC-II/peptide (pMHC-II) complexes on the surface of antigen-presenting cells (APCs). LAG-3 has been shown to selectively bind pMHC-II complexes that are considered stable after H2-DM has replaced the class-II-associated invariant chain peptide or unstable peptides by peptides with high affinity for the peptide-binding groove of MHC-II. LAG-3 is able to bind these stable pMHC-II complexes with a stronger affinity than CD4[15][18][19][20][21]. Several other ligands interact with LAG-3, including Galectin-3 (Gal-3), liver sinusoidal endothelial cell lectin (LSECtin, CLEC4G) and fibrinogen like protein 1 (FGL-1) (Figure 1).
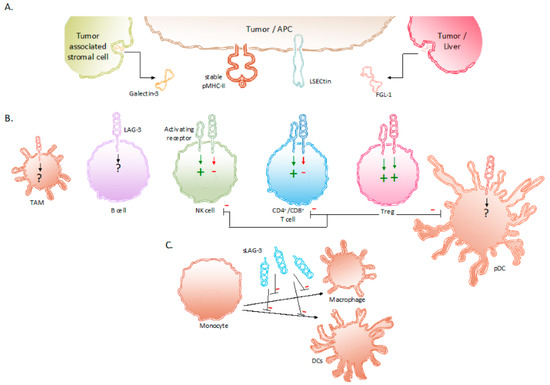
Figure 1. Expression of LAG-3 and its ligands in the TME. (A) Binding partners reported to associate with LAG-3 are stable pMHC-II complexes, Gal-3, LSECtin and FGL-1. These LAG-3 ligands are expressed on APCs, tumor cells and tumor-associated stromal cells. (B) Expression of LAG-3 on different immune cells and its influence on their effector functions. (C) sLAG-3 inhibiting the differentiation of monocytes towards macrophages or DCs. Abbreviations: APC, antigen-presenting cell; FGL-1, fibrinogen-like protein 1; Gal-3, Galectin-3; LSECtin, liver sinusoidal endothelial cell lectin; NK, natural killer; pDC, plasmacytoid dendritic cell; pMHC-II, peptide major histocompatibility complex class II; sLAG-3, soluble LAG-3; TAM, tumor-associated macrophage; TME, tumor microenvironment; Treg, regulatory T cell.
LSECtin is mainly expressed in the liver and has been found on the surface of tumor cells, such as human melanoma cells, as an engendered mechanism of immune escape[22][23]. Gal-3 is a soluble molecule secreted by a variety of tumor cells and tumor-associated stromal cells. It interacts with LAG-3 and was observed to reduce the frequency of CD8+ T cells producing interferon gamma (IFN-γ) in the TME [24][25]. The fourth ligand of LAG-3, FGL-1, has been identified more recently. FGL-1 is produced and secreted by tumor cells and hepatocytes. Preclinical work showed that FGL-1 reduced the production of IL-2 by a T-cell line, when interacting with LAG-3 on the T-cell surface [26].
3. Expression of LAG-3 and Its Regulation in Tumor-Associated Immune Cells
As illustrated in Figure 1, the expression of LAG-3 in the TME has been observed on TILs[27], in particular CD4+ and CD8+ T cells[14][28], including regulatory T cells (Tregs)[29]; NKT cells[30]; B cells[31]; NK cells[32] as well as on plasmacytoid dendritic cells (pDCs)[33][34] and tumor-associated macrophages (TAMs)[35][36].
It has been shown that LAG-3 expression on CD4+ and CD8+ T cells, even NKT cells, is induced upon continued antigen stimulation further stimulated through exposure to IFN-γ, IL-2, IL-7, and/or IL-12[20][30][37][38]. Several transcriptional regulators have been implicated in the regulation of LAG-3 expression and T-cell exhaustion, which is the state of most TILs. These are thymocyte selection-associated high mobility group box protein (TOX), nuclear factor of activated T cells (NFAT), and nuclear receptor subfamily 4, group A (NR4A)[39][40][41][42][43][44]. LAG-3 expression is further regulated through its trafficking in cells[45]. In addition, proteolytic cleavage by ADAM10 and ADAM17, two metalloproteases, regulates LAG-3 levels on the T-cell surface, allowing T-cell activation in the initiation phase of the immune response[46]. However, sustained antigen stimulation forces LAG-3 expression on the cell surface, leading to the loss of T-cell effector functions and the development of an exhausted state.
Several Treg-populations have been characterized by high and constitutive expression of LAG-3[47][48][49]. These include CD4+ Foxp3+ Tregs, IL-10 producing Tr1 cells as well as IL-10 and transforming growth factor beta (TGF-β)3 producing CD4+ Foxp3- Tregs. It has been hypothesized that this constitutive LAG-3 expression is due to continuous TCR signaling by self-antigens, which is recognized by these Treg-populations, implicating the same transcriptional regulators, as described for CD4+ and CD8+ T cells with an effector function[50]. Moreover, early growth response gene 2 (EGR2) has been described as a critical regulator of LAG-3 expression in CD4+ Foxp3- Tregs[29].
The expression of LAG-3 on B cells has not been elucidated on the molecular level. However, it has been shown to depend on activated T cells[31]. B cells have been shown to express LAG-3 when cultured with anti-CD3 antibody activated T cells, however, not when stimulated with antibodies that bind the B-cell receptor and CD40, mimicking interaction with CD4+ TH cells. Further analysis of the mechanisms leading to LAG-3 acquisition on the B-cell surface has revealed the need for a soluble factor to induce endogenous LAG-3 expression in B cells with IL-6 being a good candidate. It has been further shown using LAG-3 knock-out cells that LAG-3 detected on B cells upon co-culture with activated T cells can further be acquired through the absorption of LAG-3 from activated T cells.
The regulation of LAG-3 expression on cells of the innate immune system, including NK cells, pDCs, and macrophages, has yet to be elucidated. Although some hints to the signals that can induce LAG-3 expression on NK cells and pDCs are available, the molecular mechanisms are not yet described. LAG-3 expression has been induced on NK cells using IL-12[51][52][53]. Moreover, a subpopulation of NK cells, more specifically NKG2C+ NK cells, have been shown to express LAG-3 in response to interaction with an NKG2C agonist and IL-15[32]. These cells further showed increased PD-1 expression, suggesting that LAG-3 and PD-1 expression coincide in this NK cell population, similar as in T cells. LAG-3 expression has been shown on a subset of pDCs in healthy individuals as well as on pDCs in melanoma patients with a clear enrichment in LAG-3+ pDCs in melanoma-invaded lymph nodes and in cutaneous melanoma metastasis. Moreover, it was shown that stimuli such as IL-3 and CpG DNA could stimulate LAG-3 expression on these cells[34].
4. Signaling Induced by LAG-3 and Its Net Effect
Thirty years after its discovery, the exact signaling pathway of LAG-3 remains unknown. Compared to other ICPs such as PD-1 and CTLA-4, LAG-3 does not have immunoreceptor tyrosine-based inhibitory motifs (ITIM) present in its cytoplasmatic tail. It has been shown that the inhibitory effect of LAG-3 is not caused by competitive pMHC-II binding with CD4[21][28]. Instead, Workman et al. reported an intracellular “KIEELE” sequence in the intracellular part of LAG-3 on CD4+ T cells that is necessary to transduce distinctive yet undetermined inhibitory signals[19]. This finding was recently contradicted in a study reporting that LAG-3 elicits inhibitory mechanisms through an FXXL motif present in the membrane-proximal regions in cooperation with a C-terminal EX repeat[54]. The latter is found to interact with a so-called LAG-3-associated protein (LAP)[55]. The inhibitory effect of LAG-3 was lost upon deletion of the EX repeat and when mutations were induced in the FXXL sequence[54]. Although the molecular mechanisms exploited by LAG-3 to install inhibitory signals are slowly being uncovered, there is still much to learn about LAG-3 and how its atypical cytoplasmic motifs interact with cytoplasmic signaling proteins.
The biological activity of LAG-3 is most intriguing. LAG-3 has been shown to transduce inhibitory signals on activated CD8+ T cells even though the activation of CD8+ T cells is not driven by peptide presentation in MHC-II. Nonetheless, the inhibition of CD8+ T-cell activation has been shown to be induced by APCs that express high amounts of pMHC-II in addition to pMHC-I [21]. Moreover, together with the co-expression of PD-1, the anti-tumor immunity of CD8+ T cells could be abolished through the interaction with LAG-3’s other ligands i.e., LSECtin, FGL-1, and Gal-3 found in the TME [23][24][26][56]. Furthermore, Tregs present in the TME are renowned to weaken cancer-specific immune responses through the downregulation of inflammatory cytokines and the upregulation of suppressor activity. The role of LAG-3 has been shown to be essential in supporting Treg activity[47]. Research in non-small-cell lung cancer (NSCLC) patients show elevated LAG-3 expression on Tregs residing in the tumor compared to Tregs found in peripheral blood and normal tissues[57][58]. The surface expression of LAG-3 on Tregs has been shown to increase the secretion of immune suppressive cytokines, such as IL-10 and TGF-β[57][58]. These cytokines elicit inhibitory effects on the activity of CD8+ T cells, NK cells, and DCs. Furthermore, Tregs directly inhibit pDCs through LAG-3–pMHC-II interactions. These have been shown to initiate suppressive pathways, which hamper the proliferation and maturation of DCs[59]. Moreover, of all DC subsets, pDCs constitutively express LAG-3, which in turn negatively regulates their activation, intrinsic physiology, and extrinsic interplay with T cells[33]. Consequently, tumor-infiltrating pDCs expressing LAG-3 have been shown to contribute to an anti-inflammatory environment in melanoma patients [34]. The direct role of LAG-3 expressed on activated NK cells is still not fully understood. Although upregulated on human NK cells in response to IL-12, the blockade of LAG-3 on NK cells has shown no specific influence on their functionality[51][52][53]. However, on NKT cells expressing both NK receptors and T cells receptors, LAG-3 has shown to downregulate their proliferation[30]. As mentioned above, the expression of LAG-3 on B cells has been shown to be T-cell dependent[31]. More recently, Lino et al. identified a plasma B cell subset, selectively expressing LAG-3, with immune suppressing activity through the production of IL-10[60]. Additionally, digital spatial protein analysis demonstrated the expression of LAG-3 in TAMs[35]. Although its role is still not fully understood, it can be speculated that LAG-3 expression on TAMs contributes to their tumor-promoting function, as suggested by the association of co-expression of CD163 and LAG-3 with poor clinicopathological indexes in melanoma[36] and metastatic ovarian cancer [61]. As mentioned above, LAG-3 can be cleaved from the cell surface by ADAM10 and ADAM17 and form soluble LAG-3 (sLAG-3)[46]. sLAG-3 regulates immune responses in the TME and periphery, for example by inhibiting the differentiation of monocytes to macrophages or DCs (Figure 1)[62][63][64].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010075
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Awad, R.M.; De Vlaeminck, Y.; Maebe, J.; Goyvaerts, C.; Breckpot, K. Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front. Immunol. 2018, 9, 1977.
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-cell Function to Promote Tumoral Immune Escape. Cancer Res. 2011, 72, 917–927.
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1–specific CD8+T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880.
- Broos, K.; Lecocq, Q.; Xavier, C.; Bridoux, J.; Nguyen, T.T.; Corthals, J.; Schoonooghe, S.; Lion, E.; Raes, G.; Keyaerts, M.; et al. Evaluating a Single Domain Antibody Targeting Human PD-L1 as a Nuclear Imaging and Therapeutic Agent. Cancers 2019, 11, 872.
- Nguyen, L.T.; Ohashi, P.S. Clinical blockade of PD1 and LAG3—Potential mechanisms of action. Nat. Rev. Immunol. 2015, 15, 45–56.
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J. Clin. Oncol. 2017, 35, 9520.
- European Society for Medical Oncology. ESMO 2017 Congress Scientific Meeting Report [Internet]. 2007. Available online: https://oncologypro.esmo.org/content/download/126251/2385263/file/ESMO-2017-Congress-Scientific-Meeting-Report.pdf (accessed on 3 November 2020).
- Broos, K.; Lecocq, Q.; Raes, G.; Devoogdt, N.; Keyaerts, M.; Breckpot, K. Noninvasive imaging of the PD-1:PD-L1 immune checkpoint: Embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics 2018, 8, 3559–3570.
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.; Carvajal, R.; Sosman, J.; Atkins, M.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454.
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I Study of Single-Agent Anti–Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J. Clin. Oncol. 2010, 28, 3167–3175.
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501.
- Tang, H.; Liang, Y.; Anders, R.A.; Taube, J.M.; Qiu, X.; Mulgaonkar, A.; Liu, X.; Harrington, S.M.; Guo, J.; Xin, Y.; et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J. Clin. Investig. 2018, 128, 580–588.
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405.
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721.
- Kim, S.-S.; Kim, S.-H.; Kang, H.-S.; Chung, H.Y.; Choi, I.; Cheon, Y.-P.; Lee, K.H.; Lee, D.-M.; Park, J.; Lee, S.Y.; et al. Molecular cloning and expression analysis of pig lymphocyte activation gene-3 (LAG-3; CD223). Veter-Immunol. Immunopathol. 2010, 133, 72–79.
- Dijkstra, J.M.; Somamoto, T.; Moore, L.; Hordvik, I.; Ototake, M.; Fischer, U. Identification and characterization of a second CD4-like gene in teleost fish. Mol. Immunol. 2006, 43, 410–419.
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749.
- Workman, C.J.; Rice, D.S.; Dugger, K.J.; Kurschner, C.; Vignali, D.A.A. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur. J. Immunol. 2002, 32, 2255–2263.
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836.
- Maruhashi, T.; Okazaki, I.-M.; Sugiura, D.; Takahashi, S.; Maeda, T.K.; Shimizu, K.; Okazaki, T. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol. 2018, 19, 1415–1426.
- Liu, W.; Tang, L.; Zhang, G.; Wei, H.; Cui, Y.; Guo, L.; Gou, Z.; Chen, X.; Jiang, D.; Zhu, Y.; et al. Characterization of a novel C-type lectin-like gene, LSECtin: Demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. J. Biol. Chem. 2004, 279, 18748–18758.
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-cell Responses. Cancer Res. 2014, 74, 3418–3428.
- Kouo, T.S.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.D.; Jaffee, E.M. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423.
- Ruvolo, P.P. Galectin 3 as a guardian of the tumor microenvironment. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1863, 427–437.
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, Y.; Zhu, G.; Yin, W.; Zheng, L.; et al. Faculty Opinions recommendation of Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.
- Demeure, C.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell–cell contacts. Eur. J. Cancer 2001, 37, 1709–1718.
- Okazaki, T.; Okazaki, I.-M.; Wang, J.; Sugiura, D.; Nakaki, F.; Yoshida, T.; Kato, Y.; Fagarasan, S.; Muramatsu, M.; Eto, T.; et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J. Exp. Med. 2011, 208, 395–407.
- Okamura, T.; Fujio, K.; Shibuya, M.; Sumitomo, S.; Shoda, H.; Sakaguchi, S.; Yamamoto, K. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc. Natl. Acad. Sci. USA 2009, 106, 13974–13979.
- Byun, H.; Jung, W.; Lee, D.; Kim, S.; Park, C.; Chung, H.Y.; Chun, T. Proliferation of activated CD1d-restricted NKT cells is down-modulated by lymphocyte activation gene-3 signaling via cell cycle arrest in S phase. Cell Biol. Int. 2007, 31, 257–262.
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088.
- Merino, A.; Zhang, B.; Dougherty, P.; Luo, X.; Wang, J.; Blazar, B.R.; Miller, J.S.; Cichocki, F. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Investig. 2019, 129, 3770–3785.
- Workman, C.J.; Wang, Y.; El Kasmi, K.C.; Pardoll, D.M.; Murray, P.J.; Drake, C.G.; Vignali, D.A.A. LAG-3 Regulates Plasmacytoid Dendritic Cell Homeostasis. J. Immunol. 2009, 182, 1885–1891.
- Camisaschi, C.; De Filippo, A.; Beretta, V.; Vergani, B.; Villa, A.; Vergani, E.; Santinami, M.; Cabras, A.D.; Arienti, F.; Triebel, F.; et al. Alternative Activation of Human Plasmacytoid DCs In Vitro and in Melanoma Lesions: Involvement of LAG-3. J. Investig. Dermatol. 2014, 134, 1893–1902.
- Keane, C.; Law, S.C.; Gould, C.M.; Birch, S.; Sabdia, M.B.; De Long, L.M.; Thillaiyampalam, G.; Abro, E.; Tobin, J.W.; Tan, X.; et al. LAG3: A novel immune checkpoint expressed by multiple lymphocyte subsets in diffuse large B-cell lymphoma. Blood Adv. 2020, 4, 1367–1377.
- Kim, Y.J.; Won, C.H.; Lee, M.W.; Choi, J.-H.; Chang, S.-E.; Lee, W.J. Correlation Between Tumor-Associated Macrophage and Immune Checkpoint Molecule Expression and Its Prognostic Significance in Cutaneous Melanoma. J. Clin. Med. 2020, 9, 2500.
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, aah3374.
- Wu, J.; Zhang, H.; Shi, X.; Xiao, X.; Fan, Y.; Minze, L.J.; Wang, J.; Ghobrial, R.M.; Xia, J.; Sciammas, R.; et al. Ablation of Transcription Factor IRF4 Promotes Transplant Acceptance by Driving Allogenic CD4+ T Cell Dysfunction. Immunity 2017, 47, 1114–1128.e6.
- Chen, J.; López-Moyado, I.F.; Seo, H.; Lio, C.W.J.; Hempleman, L.J.; Sekiya, T.; Yoshimura, A.; Scott-Browne, J.P.; Rao, A. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019, 567, 530–534.
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218.
- Liu, X.; Wang, Y.; Lu, H.; Li, J.; Yan, X.; Xiao, M.; Huang, R.; Wu, J.; Zhao, Q.; Wu, Q.; et al. Faculty Opinions recommendation of Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 2019, 567, 525–529.
- Martinez, G.J.; Pereira, R.M.; Äijö, T.; Kim, E.Y.; Marangoni, F.; Pipkin, M.E.; Togher, S.; Heissmeyer, V.; Zhang, Y.C.; Crotty, S.; et al. The Transcription Factor NFAT Promotes Exhaustion of Activated CD8 + T Cells. Immunity 2015, 42, 265–278.
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415.
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.J.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274.
- Bae, J.; Lee, S.J.; Park, C.-G.; Lee, Y.S.; Chun, T. Trafficking of LAG-3 to the Surface on Activated T Cells via Its Cytoplasmic Domain and Protein Kinase C Signaling. J. Immunol. 2014, 193, 3101–3112.
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; A Black, R.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26, 494–504.
- Rouse, B. Faculty Opinions recommendation of Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513.
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746.
- Okamura, T.; Sumitomo, S.; Morita, K.; Iwasaki, Y.; Inoue, M.; Nakachi, S.; Komai, T.; Shoda, H.; Miyazaki, J.-I.; Fujio, K.; et al. TGF-β3-expressing CD4+CD25−LAG3+ regulatory T cells control humoral immune responses. Nat. Commun. 2015, 6, 6329.
- Li, M.O.; Rudensky, A.Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016, 16, 220–233.
- Huard, B.; Tournier, M.; Triebel, F. LAG-3 does not define a specific mode of natural killing in human. Immunol. Lett. 1998, 61, 109–112.
- Kritikou, J.S.; Dahlberg, C.I.M.; Baptista, M.A.P.; Wagner, A.K.; Banerjee, P.P.; Gwalani, L.A.; Poli, C.; Panda, S.K.; Kärre, K.; Kaech, S.M.; et al. IL-2 in the tumor microenvironment is necessary for Wiskott-Aldrich syndrome protein deficient NK cells to respond to tumors in vivo. Sci. Rep. 2016, 6, 30636.
- Sun, H.; Sun, C.; Xiao, W. Expression regulation of co-inhibitory molecules on human natural killer cells in response to cytokine stimulations. Cytokine 2014, 65, 33–41.
- Maeda, T.K.; Sugiura, D.; Okazaki, I.-M.; Maruhashi, T.; Okazaki, T. Atypical motifs in the cytoplasmic region of the inhibitory immune co-receptor LAG-3 inhibit T cell activation. J. Biol. Chem. 2019, 294, 6017–6026.
- Iouzalen, N.; Andreae, S.; Hannier, S.; Triebel, F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. Eur. J. Immunol. 2001, 31, 2885–2891.
- Bos, R.; Marquardt, K.L.; Cheung, J.; Sherman, L.A. Functional differences between low- and high-affinity CD8+T cells in the tumor environment. OncoImmunology 2012, 1, 1239–1247.
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 Expression Defines a Subset of CD4+CD25highFoxp3+ Regulatory T Cells That Are Expanded at Tumor Sites. J. Immunol. 2010, 184, 6545–6551.
- Wei, T.; Zhang, J.; Qin, Y.; Wu, Y.; Zhu, L.; Lu, L.; Tang, G.; Shen, Q. Increased expression of immunosuppressive molecules on intratumoral and circulating regulatory T cells in non-small-cell lung cancer patients. Am. J. Cancer Res. 2015, 5, 2190–2201.
- Liang, B.; Workman, C.; Lee, J.; Chew, C.; Dale, B.M.; Colonna, L.; Flores, M.; Li, N.; Schweighoffer, E.; Greenberg, S.; et al. Regulatory T Cells Inhibit Dendritic Cells by Lymphocyte Activation Gene-3 Engagement of MHC Class II. J. Immunol. 2008, 180, 5916–5926.
- Lino, A.C.; Dang, V.D.; Lampropoulou, V.; Welle, A.; Joedicke, J.; Pohar, J.; Simon, Q.; Thalmensi, J.; Baures, A.; Flühler, V.; et al. LAG-3 Inhibitory Receptor Expression Identifies Immunosuppressive Natural Regulatory Plasma Cells. Immunity 2018, 49, 120–133.e9.
- Hensler, M.; Kasikova, L.; Fiser, K.; Rakova, J.; Skapa, P.; Laco, J.; Lanickova, T.; Pecen, L.; Truxova, I.; Vosahlikova, S.; et al. M2-like macrophages dictate clinically relevant immunosuppression in metastatic ovarian cancer. J. Immunother. Cancer 2020, 8, e000979.
- Lienhardt, C.; Azzurri, A.; Amedei, A.; Fielding, K.; Sillah, J.; Sow, O.Y.; Bah, B.; Benagiano, M.; Diallo, A.; Manetti, R.; et al. Active tuberculosis in Africa is associated with reduced Th1 and increased Th2 activity in vivo. Eur. J. Immunol. 2002, 32, 1605–1613.
- Triebel, F. LAG-3: A regulator of T-cell and DC responses and its use in therapeutic vaccination. Trends Immunol. 2003, 24, 619–622.
- Annunziato, F.; Manetti, R.; Tomasévic, I.; Giudizi, M.G.; Biagiotti, R.; Giannò, V.; Germano, P.; Mavilia, C.; Maggi, E.; Romagnani, S. Expression and release of LAG-3-encoded protein by human CD4+ T cells are associated with IFN-gamma production. FASEB J. 1996, 10, 769–776.