Acute myeloid leukemia (AML) is a heterogenous disease with multiple sub-types which are defined by different somatic mutations that cause blood cell differentiation to go astray. One of the best-studied AML-subtypes is the t(8;21) AML which carries a translocation fusing sequences encoding the DNA-binding domain of the hematopoietic master regulator RUNX1 to the ETO gene.
- Acute myeloid leukemia (AML)
- Core binding factor AML
- Fusion protein RUNX1/ETO (RUNX1/RUNX1T1)
- epigenetic reprogramming
- gene regulatory network reprogramming
- targeted therapy for AML
1. Introduction
Acute Myeloid Leukemia (AML) is a heterogeneous disease characterised by proliferation of neoplastic cells with impaired myeloid differentiation. Distinct classes of driver mutations alter the differentiation trajectory and epigenetic landscape differently and instead of following a normal trajectory, malignant cells adopt new identities distinct from normal cells which are maintained by specific gene regulatory networks that drive common and subtype-specific AML signalling and metabolic pathways. In adults, it is mainly a disease of the elderly and is rarely cured due to the high relapse rates. With few exceptions, for most AML subtypes the only treatment option is standard chemotherapy using regimes that have little changed in decades and which are often not an option for elderly patients. In the last years, research has therefore turned away from treating AML as a single disease entity and has undergone a shift towards understanding each subtype in molecular detail, in the hope to be able to interfere with leukemic growth in a specific fashion. These efforts have led to the development of highly effective specific inhibitors in other types of leukemia by targeting the oncoproteins causing APL (PML/RAR; ATRA) and CML (BCR/ABL; imatinib), but have failed to effectively target others in AML, such as the FLT3-ITD growth factor receptor whose mutation causes a particularly aggressive type of AML. Such failures have highlighted our lack of understanding of how different mutant proteins reprogram normal into malignant cells and which factors are required for their survival. One type of AML, the t(8;21) and its oncogenic fusion protein, RUNX1/ETO have been studied for several decades at multiple levels and serves as a paradigm of how specific mutations reprogram the epigenome.
2. The Chromosomal Rearrangement t(8;21) Gives Rise to a Fusion Protein
Chromosomal translocations are a major initiator of cell transformation in hematological malignancies, and recurrently involve transcription factor genes that regulate normal hematopoiesis [1][2]. A highly prevalent rearrangement found in AML occurs in the gene loci encoding members of the core-binding factor (CBF) complex, consisting of a heterodimeric transcription factor complex composed of a member of the RUNX DNA binding factor family (CBFα/ AML1/ RUNX1) and a non-DNA binding partner termed CBFβ [3]. Chromosomal rearrangements target both components of the CBF complex: the translocations t(8;21)(q22;q22) and t(16;21)(q24;q22) fuse part of RUNX1 to members of the ETO (Eight Twenty-one) family [4][5]. CBF leukemia accounts for nearly 25% of pediatric AML cases, with t(8;21) alone being present in 15% of all cases. Its incidence decreases in older patients to 5% [6]. CBF leukemias are considered as good-prognosis AML, however, older patients are often subject to chemotherapy failure and relapse [7].
During embryogenesis, the transcription factor RUNX1 is essential to generate hematopoietic stem and progenitor cells (HSPCs). Depletion of RUNX1 at this stage is lethal in mice due to a total lack of hematopoiesis [8]. However, in adult hematopoiesis, RUNX1 is not essential for the maintenance of self-renewal capacity of HSCs [9]. ETO (also known as RUNX1T1) is highly expressed in neurons, but its cellular functions in humans have been mainly identified as part of the RUNX1/ETO complex in AML. ETO-interactors include co-repressor complexes suggesting that this protein is a transcriptional repressor that is located in nuclear bodies [10][11]. Although highly expressed in the adult brain, insertional mutagenesis in the murine embryo leads to massive defects in gastrointestinal development, with no detected abnormality in hematopoietic system [12]. Thus, the precise function of ETO in various cellular contexts remains to be fully characterised.
The t(8;21) translocation fuses the N-terminal DNA binding Runt Homology Domain (RHD) domain of RUNX1 to the almost complete ETO protein, creating a chimeric protein with 752 amino acids (Figure 1). The fusion protein maintains its ability to interact via its RHD with CBFβ and with DNA. ETO contributes four Nervy Homology Regions (NHR1-4) to the fusion protein. NHR1 has sequence homology to TATA-binding protein-associated factors and seems to be dispensable for gene repression by RUNX1/ETO. Nevertheless, its depletion abolishes formation of ETO nuclear bodies and suggests a role in the subcellular localization of ETO [10]. The NHR2 domain is essential for leukemogenic activity, it mediates homo and heterodimerisation with ETO members and recruits the NCoR/SIN3A co-repressor together with histone deacetylases (HDACs) [13][14][15]. Tetramerisation of the NHR2 domain itself is also essential for the leukemogenic activity of RUNX1/ETO, as mono- or dimeric fusion proteins do not efficiently bind DNA. Consequently, depletion of the NHR2 domain reverts the repressive effects of RUNX1/ETO on myeloid differentiation, and interfering with the oligomerisation by peptides abrogates the effect of RUNX1/ETO on leukemic self-renewal [16][17]. NHR4 recruits, SMRT (Silencing Mediator of Retinoic Acid and Thyroid Hormone Receptors) and SIN3, class I HDACs via nuclear receptor corepressor (NCOR) [11]. NHR3 assists NHR4 to interact with NCOR. However, binding to NCoR by NHR3 and NHR4 is not sufficient to induce maximal transcriptional repression [18].
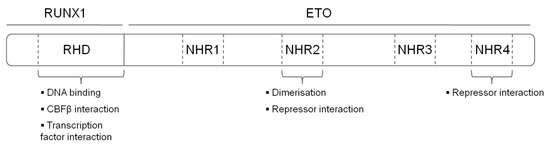
Figure 1. Structure and functional domains of the RUNX1/ETO fusion protein. RHD—Runt homology domain, NHR—nervy homology region.
3. RUNX1/ETO on Its Own Does Not Cause AML
Mouse models have been instrumental in elucidating the function of RUNX1/ETO in blood cell development and differentiation. One of the earliest RUNX1/ETO mouse models inserted a RUNX1/ETO fusion cDNA into one allele of the murine Runx1 locus which caused an embryonic lethal phenotype. Definitive fetal liver-derived hematopoiesis was perturbed together with lethal hemorrhages indicating that endothelial development was affected as well [19]. This phenotype strongly resembled that found in homozygous Runx1 knock-out mice [20]. It was, therefore, suggested that RUNX1/ETO functions as a dominant-negative version of RUNX1.
To bypass the embryonic lethality of the knock-in model, conditional transgenic mouse models were developed that express RUNX1/ETO in adult bone marrow progenitors either using CRE/LoxP mediated recombination [21][22] or express the fusion protein under the control of the tetracycline (Tet) promoter [23]. Another inducible murine model carried RUNX1/ETO as a conditional and reversible transgene [24]. An important result from these studies was that while cells showed aberrant growth and differentiation phenotypes, RUNX1/ETO expression did not cause a full-blown AML, demonstrating that the t(8;21) mutation needs to cooperate with other mutations. This idea was confirmed when RUNX1/ETO expressing mice did succumb to AML development after exposure to the genotoxic drug N-ethyl-N-nitrosourea [22][25]. These experimental findings are in line with the detection of this translocation in Guthrie spots of newborns that were diagnosed with AML years later as adults [26]. Furthermore, the RUNX1/ETO transcript can be still found in the early hematopoietic stem or progenitor cells from AML patients in complete remission suggesting the existence of a RUNX1/ETO-positive pre-leukemic state [27]. Last, but not least, the majority of t(8;21) AML patients present with additional mutations that drive leukemic growth such as activating mutations in receptor tyrosine kinases which are present in at least 30% of patients with AML. When investigating the cooperating effect of expressing mutated kinases (such as mutant FLT3, RAS or KIT) together with by retrovirally transducing murine bone marrow cells with RUNX1/ETO into lethally irradiated syngeneic mice, all animals developed AML [28][29][30][31][32].
4. Molecular aspects of RUNX1/ETO function
The leukemic phenotype of t(8;21) AML cells is strictly dependent on the presence of RUNX1/ETO. Knock down of RUNX1/ETO transcript by siRNAs targeting the fusion region decreases the self-renewal capacity and induces differentiation [33]. Histone deacetylation and gene repression via ETO mediated recruitment of co-repressors and HDACs have been suggested as the main mechanism for leukemogenic activities of RUNX1/ETO [34]. Thus, fusion of ETO provides a docking site to introduce novel interaction partners to RUNX1 target regions and creates a shift of the balance of activation and repression [13]. RUNX1/ETO functions as part of a stable transcription factor complex termed AML1/ETO-containing transcription factor complex (AETFC) [13]. Pull down experiments with t(8;21) cells followed by mass spectrometry-based identification of the complex showed that RUNX1/ETO complex is enriched in RUNX1/ETO, CBFβ, FLI1/ERG, LMO2, LYL1, HDAC 1 and 2, p300, number of SRS and RBM splicing factors. Leukaemogenesis of t(8;21) is therefore not solely dependent on expression of RUNX1/ETO but its impact is the consequence of its interference with the combinatorial interplay between different hematopoietic transcriptional regulators.
5. System wide studies on aberrant chromatin programming by RUNX1/ETO
The use of system-wide techniques such as chromatin immunoprecipitation (ChIP) assays, DNaseI hypersensitive site mapping and RNA-Sequencing has been essential to obtain global insights into how RUNX1/ETO blocks cell differentiation and leads to extensive changes in gene regulation. RUNX1/ETO affects the entire chromatin landscape including chromatin accessibility, hematopoietic regulator binding and histone modification patterns in a dynamic fashion [34][35][36]. The mechanism behind the reorganisation of chromatin is primarily due to redistribution and displacement of RUNX1 by RUNX1/ETO (56-58) which sets up an aberrant gene regulatory network that is unique to t(8;21) leukemia [37]. RUNX1/ETO is localised predominately to open chromatin, enriched for ETS and RUNX binding sites and regulates the balance between the p300 co-activator, HDACs and acetylated histones [34][36][38].
The presence of repressive NHR domains in the ETO protein, together with the strong binding preference of RUNX1/ETO to active chromatin sites have historically led to RUNX1/ETO being considered as a repressor protein [39][40]. More recently, there is a growing body of evidence from global studies that RUNX1/ETO interferes with both the activating and repressive actions of RUNX1 with crucial knock-on effects in the gene regulatory networks. Gene regulation by RUNX1/ETO at the chromatin level is therefore a complex process, as a consequence of mixed interactions between co-activators, co-repressors and other transcription factors.
6. Therapeutic targeting of RUNX1/ETO
RUNX1/ETO-positive AML has in general a favourable clinical prognosis with overall survival ranging from 65% from younger adults to 75% in children but its rate of relapse is high [41]. With the exception of mylotarg, an anti-CD33 antibody drug conjugate, targeted drugs have so far not contributed to the outcome. Instead and regardless of the molecular subtype, chemotherapy has remained the cornerstone of treatment for AML for over 25 years. Induction and remission phases include the use of high dose nucleoside analogues (e.g cytarabine), combined with anthracyclines (idarubicin or daunorubicin) [42]. Recent clinical trials may escalate the dose or duration to optimize the protocols and induce complete remission of the disease with minimal risk of relapse [43]. However, such dose escalation may not be applicable for majority of elderly patients. Moreover, due to their longer life expectancy, children are particularly affected by long-term consequences of chemotherapy such as irreversible cardiotoxicity caused by anthracyclines urging the need for targeted therapies, urging the need for targeted therapies.
Leukemic fusion genes such as RUNX1/ETO constitute ideal targets for leukemia therapy. Since RUNX1/ETO initiates initiates leukaemogenesis, all leukemic and pre-leukemic cells in a patient will harbour it. Furthermore, since it is still an essential driver of leukemia,
perturbing its expression or function will affect leukemic progression. Current approaches to directly target RUNX1/ETO comprises the use of siRNAs targeting the fusion site of its mRNA or peptide-mediated inhibition of homo- or hetero-oligomerisation with itself and other ETO family members via its NHR3 domain [44][45][46]. Both approaches yielded very similar outcomes in cell lines with loss of self-renewal and facilitation of myeloid differentiation. Both peptides and siRNAs have poor pharmacokinetic properties complicating their therapeutic application. However, the field of therapeutic siRNA delivery is intensively investigated and the approval of two siRNA formulations for the treatment of liver-associated pathologies raises the hope that similar breakthroughs might be achievable for the therapeutic targeting of leukemic fusion transcripts. Genes that are activated by RUNX1/ETO expression were screened for their relevance in the RUNX1/ETO-driven leukemic programme by performing a targeted RNAi screen both in tissue culture and in vivo [47]. This screen identified CCND2 as an essential downstream target for RUNX/1ETO. RUNX1/ETO drives cell cycle progression in the G1 phase by also sustains the expression of its partner protein CDK6. Knockdown of CCND2 in RUNX1/ETO-expressing AML cells yielded a gene signature that was highly similar to that of cells treated with the clinically approved CDK4/6 inhibitor palbociclib. Further evaluation both ex vivo in patient-derived AML cells and in vivo in immunodeficient mice transplanted with RUNX1/ETO-expressing AML cells demonstrated a high sensitivity of this AML subtype to palbociclib inhibition.
7. Outlook
The increased understanding of t(8;21) AML biology is now also increasingly translated into novel, more leukemia-specific therapeutic concepts. In particular, targeting leukemic oncoproteins and components of the gene regulatory networks maintaining the leukemic phenotype is now rapidly becoming a reality [48]. the next logical steps of their clinical evaluation will be challenging as t(8;21) has a good response to existing chemotherapy protocols. Especially in the paediatric setting, this creates a bottleneck for clinical testing due to the low number of refractory or relapsing patients being available for testing these new concepts. In spite of these issues: It is important to note that chemotherapy is highly genotoxic. Children and young adults will therefore be more affected by the long-term consequences of chemotherapy giving rise to secondary cancers, thus justifying clinical efforts to improve their quality of life by establishing more precise and milder approaches to treat t(8;21) AML. A successful precision medicine approach - in the true meaning of the word, not just “which toxic treatment is going to work for this patient” - would not only benefit our patients, but would serve again as a paradigm for a cure with minimal long-term burdens.
This entry is adapted from the peer-reviewed paper 10.3390/cells9122681
References
- Mrózek, K.; Bloomfield, C.D. Clinical significance of the most common chromosome translocations in adult acute myeloid leukemia. J. Natl. Cancer Inst. Monogr. 2008, 2008, 52–57.
- Wiemels, J. Chromosomal translocations in childhood leukemia: Natural history, mechanisms, and epidemiology. J. Natl. Cancer Inst. Monogr. 2008, 2008, 87–90.
- Speck, N.A.; Stacy, T.; Wang, Q.; North, T.; Gu, T.-L.; Miller, J.; Binder, M.; Marin-Padilla, M. Core-binding factor: A central player in hematopoiesis and leukemia. Cancer Res. 1999, 59, 1789s–1793s.
- Miyoshi, H.; Shimizu, K.; Kozu, T.; Maseki, N.; Kaneko, Y.; Ohki, M. t (8; 21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc. Natl. Acad. Sci. USA 1991, 88, 10431–10434.
- de Bruijn, M.F.; Speck, N.A. Core-binding factors in hematopoiesis and immune function. Oncogene 2004, 23, 4238–4248.
- Sood, R.; Kamikubo, Y.; Liu, P. Role of RUNX1 in hematological malignancies. Blood J. Am. Soc. Hematol. 2017, 129, 2070–2082.
- Solh, M.; Yohe, S.; Weisdorf, D.; Ustun, C. Core-binding factor acute myeloid leukemia: Heterogeneity, monitoring, and therapy. Am. J. Hematol. 2014, 89, 1121–1131.
- Yzaguirre, A.D.; de Bruijn, M.F.; Speck, N.A. The role of Runx1 in embryonic blood cell formation. In RUNX Proteins in Development and Cancer; Springer: Berlin/Heidelberg, Germany, 2017; pp. 47–64.
- Link, K.A.; Chou, F.S.; Mulloy, J.C. Core binding factor at the crossroads: Determining the fate of the HSC. J. Cell. Physiol. 2010, 222, 50–56.
- Odaka, Y.; Mally, A.; Elliott, L.T.; Meyers, S. Nuclear import and subnuclear localization of the proto-oncoprotein ETO (MTG8). Oncogene 2000, 19, 3584–3597.
- Hug, B.A.; Lazar, M.A. ETO interacting proteins. Oncogene 2004, 23, 4270–4274.
- Calabi, F.; Pannell, R.; Pavloska, G. Gene Targeting Reveals a Crucial Role forMTG8 in the Gut. Mol. Cell. Biol. 2001, 21, 5658–5666.
- Sun, X.-J.; Wang, Z.; Wang, L.; Jiang, Y.; Kost, N.; Soong, T.D.; Chen, W.-Y.; Tang, Z.; Nakadai, T.; Elemento, O. A stable transcription factor complex nucleated by oligomeric AML1–ETO controls leukaemogenesis. Nature 2013, 500, 93–97.
- Li, J.; Guo, C.; Steinauer, N.; Zhang, J. New insights into transcriptional and leukemogenic mechanisms of AML1-ETO and E2A fusion proteins. Front. Biol. 2016, 11, 285–304.
- Lutterbach, B.; Westendorf, J.J.; Linggi, B.; Patten, A.; Moniwa, M.; Davie, J.R.; Huynh, K.D.; Bardwell, V.J.; Lavinsky, R.M.; Rosenfeld, M.G. ETO, a target of t (8; 21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol. Cell. Biol. 1998, 18, 7176–7184.
- Liu, Y.; Cheney, M.D.; Gaudet, J.J.; Chruszcz, M.; Lukasik, S.M.; Sugiyama, D.; Lary, J.; Cole, J.; Dauter, Z.; Minor, W. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO’s activity. Cancer Cell 2006, 9, 249–260.
- Wichmann, C.; Becker, Y.; Chen-Wichmann, L.; Vogel, V.; Vojtkova, A.; Herglotz, J.; Moore, S.; Koch, J.; Lausen, J.; Mäntele, W. Dimer-tetramer transition controls RUNX1/ETO leukemogenic activity. Blood J. Am. Soc. Hematol. 2010, 116, 603–613.
- Hildebrand, D.; Tiefenbach, J.; Heinzel, T.; Grez, M.; Maurer, A.B. Multiple regions of ETO cooperate in transcriptional repression. J. Biol. Chem. 2001, 276, 9889–9895.
- Okuda, T.; Cai, Z.; Yang, S.; Lenny, N.; Lyu, C.-j.; van Deursen, J.M.; Harada, H.; Downing, J.R. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood J. Am. Soc. Hematol. 1998, 91, 3134–3143.
- Okuda, T.; Van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330.
- Buchholz, F.; Refaeli, Y.; Trumpp, A.; Bishop, J.M. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep. 2000, 1, 133–139.
- Higuchi, M.; O’Brien, D.; Kumaravelu, P.; Lenny, N.; Yeoh, E.-J.; Downing, J.R. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t (8; 21) acute myeloid leukemia. Cancer Cell 2002, 1, 63–74.
- Rhoades, K.L.; Hetherington, C.J.; Harakawa, N.; Yergeau, D.A.; Zhou, L.; Liu, L.-Q.; Little, M.-T.; Tenen, D.G.; Zhang, D.-E. Analysis of the role of AML1-ETO in leukemogenesis, using an inducible transgenic mouse model. Blood J. Am. Soc. Hematol. 2000, 96, 2108–2115.
- Cabezas-Wallscheid, N.; Eichwald, V.; de Graaf, J.; Löwer, M.; Lehr, H.A.; Kreft, A.; Eshkind, L.; Hildebrandt, A.; Abassi, Y.; Heck, R. Instruction of haematopoietic lineage choices, evolution of transcriptional landscapes and cancer stem cell hierarchies derived from an AML1-ETO mouse model. EMBO Mol. Med. 2013, 5, 1804–1820.
- Yuan, Y.; Zhou, L.; Miyamoto, T.; Iwasaki, H.; Harakawa, N.; Hetherington, C.J.; Burel, S.A.; Lagasse, E.; Weissman, I.L.; Akashi, K. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc. Natl. Acad. Sci. USA 2001, 98, 10398–10403.
- Wiemels, J.L.; Xiao, Z.; Buffler, P.A.; Maia, A.T.; Ma, X.; Dicks, B.M.; Smith, M.T.; Zhang, L.; Feusner, J.; Wiencke, J. In utero origin of t (8; 21) AML1-ETO translocations in childhood acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2002, 99, 3801–3805.
- Shima, T.; Miyamoto, T.; Kikushige, Y.; Yuda, J.; Tochigi, T.; Yoshimoto, G.; Kato, K.; Takenaka, K.; Iwasaki, H.; Mizuno, S.; et al. The ordered acquisition of Class II and Class I mutations directs formation of human t(8;21) acute myelogenous leukemia stem cell. Exp. Hematol. 2014, 42, 955–965.e5.
- Grisolano, J.L.; O’Neal, J.; Cain, J.; Tomasson, M.H. An activated receptor tyrosine kinase, TEL/PDGFβR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 9506–9511.
- Schessl, C.; Rawat, V.P.; Cusan, M.; Deshpande, A.; Kohl, T.M.; Rosten, P.M.; Spiekermann, K.; Humphries, R.K.; Schnittger, S.; Kern, W. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J. Clin. Investig. 2005, 115, 2159–2168.
- Nick, H.J.; Kim, H.-G.; Chang, C.-W.; Harris, K.W.; Reddy, V.; Klug, C.A. Distinct classes of c-Kit–activating mutations differ in their ability to promote RUNX1-ETO–associated acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2012, 119, 1522–1531.
- Wang, Y.-Y.; Zhao, L.-J.; Wu, C.-F.; Liu, P.; Shi, L.; Liang, Y.; Xiong, S.-M.; Mi, J.-Q.; Chen, Z.; Ren, R. C-KIT mutation cooperates with full-length AML1-ETO to induce acute myeloid leukemia in mice. Proc. Natl. Acad. Sci. USA 2011, 108, 2450–2455.
- Zuber, J.; Radtke, I.; Pardee, T.S.; Zhao, Z.; Rappaport, A.R.; Luo, W.; McCurrach, M.E.; Yang, M.-M.; Dolan, M.E.; Kogan, S.C. Mouse models of human AML accurately predict chemotherapy response. Genes Dev. 2009, 23, 877–889.
- Heidenreich, O.; Krauter, J.r.; Riehle, H.; Hadwiger, P.; John, M.; Heil, G.; Vornlocher, H.-P.; Nordheim, A. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t (8; 21)-positive leukemic cells. Blood J. Am. Soc. Hematol. 2003, 101, 3157–3163.
- Ptasinska, A.; Assi, S.A.; Mannari, D.; James, S.R.; Williamson, D.; Dunne, J.; Hoogenkamp, M.; Wu, M.; Care, M.; McNeill, H.; et al. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia 2012, 26, 1829–1841.
- Saeed, S.; Logie, C.; Francoijs, K.-J.; Frigè, G.; Romanenghi, M.; Nielsen, F.G.; Raats, L.; Shahhoseini, M.; Huynen, M.; Altucci, L.; et al. Chromatin accessibility, p300, and histone acetylation define PML-RARα and AML1-ETO binding sites in acute myeloid leukemia. Blood 2012, 120, 3058–3068.
- Ptasinska, A.; Assi, S.A.; Martinez-Soria, N.; Imperato, M.R.; Piper, J.; Cauchy, P.; Pickin, A.; James, S.R.; Hoogenkamp, M.; Williamson, D. Identification of a dynamic core transcriptional network in t (8; 21) AML that regulates differentiation block and self-renewal. Cell Rep. 2014, 8, 1974–1988.
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842.
- Wang, J.; Hoshino, T.; Redner, R.L.; Kajigaya, S.; Liu, J.M. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. USA 1998, 95, 10860.
- Stengel, K.R.; Ellis, J.; Spielman, C.; Bomber, M.; Hiebert, S.W. Definition of a Small Core Transcriptional Circuit Regulated by AML1-ETO. bioRxiv 2020.
- Ray, D.; Kwon, S.Y.; Tagoh, H.; Heidenreich, O.; Ptasinska, A.; Bonifer, C. Lineage-inappropriate PAX5 expression in t(8;21) acute myeloid leukemia requires signaling-mediated abrogation of polycomb repression. Blood 2013, 122, 759–769.
- Illendula, A.; Gilmour, J.; Grembecka, J.; Tirumala, V.S.S.; Boulton, A.; Kuntimaddi, A.; Schmidt, C.; Wang, L.; Pulikkan, J.A.; Zong, H.; et al. Small Molecule Inhibitor of CBFbeta-RUNX Binding for RUNX Transcription Factor Driven Cancers. EBioMedicine 2016, 8, 117–131.
- Metz, A.; Schanda, J.; Grez, M.; Wichmann, C.; Gohlke, H. From determinants of RUNX1/ETO tetramerization to small-molecule protein–protein interaction inhibitors targeting acute myeloid leukemia. J. Chem. Inf. Modeling 2013, 53, 2197–2202.
- Wichmann, C.; Chen, L.; Heinrich, M.; Baus, D.; Pfitzner, E.; Zörnig, M.; Ottmann, O.G.; Grez, M. Targeting the oligomerization domain of ETO interferes with RUNX1/ETO oncogenic activity in t (8; 21)-positive leukemic cells. Cancer Res. 2007, 67, 2280–2289.
- Marcucci, G.; Geyer, S.; Laumann, K.; Zhao, W.; Bucci, D.; Uy, G.L.; Blum, W.; Eisfeld, A.-K.; Pardee, T.S.; Wang, E.S. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv. 2020, 4, 696–705.
- Abbas, H.A.; Alfayez, M.; Kadia, T.; Ravandi-Kashani, F.; Daver, N. Midostaurin In Acute Myeloid Leukemia: An Evidence-Based Review And Patient Selection. Cancer Manag. Res. 2019, 11, 8817.
- Lo, M.-C.; Peterson, L.F.; Yan, M.; Cong, X.; Hickman, J.H.; DeKelver, R.C.; Niewerth, D.; Zhang, D.-E. JAK inhibitors suppress t (8; 21) fusion protein-induced leukemia. Leukemia 2013, 27, 2272–2279.
- Martinez-Soria, N.; McKenzie, L.; Draper, J.; Ptasinska, A.; Issa, H.; Potluri, S.; Blair, H.J.; Pickin, A.; Isa, A.; Chin, P.S. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell 2018, 34, 626–642.e628.
- Bushweller, J.H. Targeting transcription factors in cancer-from undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624.