Neuroendocrine lung tumors comprise a range of malignancies that extend from benign tumorlets to the most prevalent and aggressive Small Cell Lung Carcinoma (SCLC). They also include low-grade Typical Carcinoids (TC), intermediate-grade Atypical Carcinoids (AC) and high-grade Large Cell Neuroendocrine Carcinoma (LCNEC). Optimal treatment options have not been adequately established: surgical resection when possible is the choice for AC and TC, and for SCLC chemotherapy and very recently, immune checkpoint inhibitors.Some mouse models have been generated based on the molecular alterations identified in genomic analyses of human tumors. With the exception of SCLC, there is a limited availability of (preclinical) models making their development an unmet need for the understanding of the molecular mechanisms underlying these diseases.
- mouse models
- lung cancer
- typical carcinoid
- atypical carcinoid
- large cell neuroendocrine lung cancer
- small cell lung carcinoma
- genetic landscape
1. Introduction
Neuroendocrine lung tumors account for approximately 20% of all lung cancers [1]. They arise in the lung unlike other neuroendocrine tumors with other anatomical body localization (mainly small intestine and rectum) [2]. Since the 2015 World Health Organization (WHO) classification they are grouped together in one category, including typical carcinoid (TC), 1.8% of lung cancers; atypical carcinoid (AC), 0.2% of the cases; large cell neuroendocrine carcinomas (LCNEC), 3%; and small cell lung carcinoma (SCLC), accounting for 15% of lung cancer cases [3][4]. They comprise a range of malignancies with markedly different traits, incidences, aggressiveness, prognosis, and management. All of them share histologic characteristics (an organoid architecture and growth pattern, with rosettes and peripheral palisading), while they differ in their proliferation count, necrosis, cell size, and nuclear features. While LCNEC and SCLC are high-grade aggressive tumors, TC and AC are well-differentiated tumors of low- and intermediate-grade malignancy, respectively, that nonetheless metastasize with a rather high frequency (10–23% of TC and 40–50% of AC) [1]. Lung low- and intermediate-grade neuroendocrine malignancies TC and AC are also known as carcinoids, which differentiate them from the WHO classification proposed for neuroendocrine tumors arising in other anatomical locations [3].
The diagnosis of neuroendocrine lung tumors relies upon histological examination and immunohistochemical detection of general markers of neuroendocrine differentiation such as chromogranin A, synaptophysin, neuroendocrine cell adhesion molecule (NCAM) and insulinoma associated protein 1 (INSM1). These markers provide a reliable profile to confirm their neuroendocrine nature [5][6][7]. However, they do not distinguish among the different subtypes of neuroendocrine tumors. Histologically, the presence of mitosis separates the groups: 2/mm2 for TC and 2–10/mm2 for AC, >10/mm2 for LCNEC and SCLC. Presence of necrosis is a feature of high-grade tumors, and LCNEC and SCLC are separated on the basis of cytological criteria (cell size and nuclear features) [3] (Figure 1). Ki-67 (a marker of proliferating cells spanning from G1 to M phase) labelling index is emerging as a discriminator between carcinoids and SCLC that helps for clinical decision making, particularly on cytology samples for non-resectable cases [5].
With regard to prognosis, patients with TC have a better prognosis than those with AC, and both have a better outcome than patients with LCNEC or SCLC that show lower survival rates (40% and 35% 5-year survival rate, respectively) [8] . Surgical resection is the treatment of choice in patients with localized TC or AC [9] and the only curative option for lung neuroendocrine tumors. When carcinoids have metastasized, they are difficult to treat due to high resistance to radiotherapy and chemotherapy. Peptide receptor radionuclide therapy (PRRT) alongside everolimus has been recently proposed for patients with advanced TC or AC [10]. For patients with LCNEC and SCLC, resection is only employed in the very few cases detected before metastatic spread at the time of diagnosis. For SCLC, chemotherapy in conjunction with radiotherapy is the standard of care, but this is only transiently effective. Immunotherapy is emerging as an alternative but only for a limited number of cases [11]. Responsiveness to SCLC chemotherapy regimens has been reported in some LCNEC series [12] but this is not a consistent finding. The optimal treatment for LCNEC has not been adequately established. There is a lack of consensus for treatment guidelines, which further complicates their management [13]. Overall, neuroendocrine lung tumors are in need of additional and novel effective (systemic and targeted) therapies.
As mentioned above, incidences vary dramatically among the different groups, and have changed over the past few years. A decrease has been reported for SCLC, while the annual incidence for carcinoids and LCNEC has increased worldwide, particularly at the advanced stages [14][15], yet they remain rare. With the exception of SCLC, which accounts for ≈15% of all lung cancers, neuroendocrine lung tumors present a limited incidence. This low incidence is one of the main reasons why a comprehensive analysis of their molecular and genomic alterations remains elusive, although recent efforts have been made in performing genomic studies in the rare neuroendocrine lung tumors (and SCLC) [16][17][18][19][20].
To circumvent this limitation, mouse models have emerged as powerful and necessary tools to delineate the development, progression, and behavior of human tumors, and to facilitate clinical application of novel therapies in patients. Another significant challenge in neuroendocrine lung cancer research is the scarcity of patient samples due to the limited incidence of the disease or owing to the fact that most diagnoses and clinical decisions are based on fine-needle aspirates or small biopsies. Therefore, mouse models, including Genetically Engineered Mouse Models (GEMMs), patient-derived and human cancer cell line xenografts, are invaluable tools to advance our understanding of the underpinnings of these diseases and to carry out translational research, identifying vulnerabilities that can potentially be targeted.
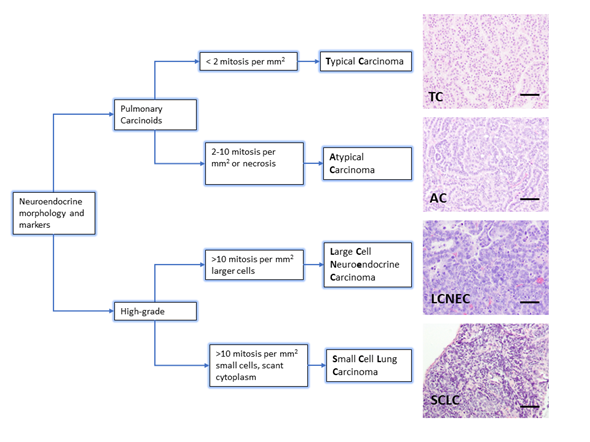
Figure 1. Lung neuroendocrine tumor types. WHO morphologic criteria for classification of lung neuroendocrine tumors. Pulmonary Carcinoids (Typical Carcinoma, TC and Atypical Carcinoma, AC) are separated from high-grade tumors (Large Cell Neuroendocrine Carcinoma, LCNEC and Small Cell Carcinoma, SCLC) according to proliferation count (mitosis). Right column shows a representative hematoxylin and eosin staining from lung tumors arisen in Genetically Engineered Mouse Models of each quoted tumor type [21][22]. Scale bars, 50 μm.
2. Genetic Landscape of Human Neuroendocrine Tumors
Human high-grade neuroendocrine tumors display a nearly universal loss of function (gene mutation, copy number loss or methylation) in either or both the tumor suppressors TP53 (which encodes for p53) and RB1 (which encodes for retinoblastoma, RB1) [23][24][25]. TP53 inactivation is a common event in neuroendocrine and non-neuroendocrine lung tumors; however, RB1 loss of function is very characteristic of the former. SCLC is the best molecularly characterized neuroendocrine tumor. The first attempts to identify SCLC subclasses based on their mutational profile [16][17][18] were not able to yield clear SCLC subtypes due to the abovementioned uniform genomic inactivation of TP53 and RB1, and to failure of other recurrent mutations (such as EP300, CREBBP, TP73, COL22A1, NOTCH1, MLL) to show reliable co-occurrence or mutual exclusivity. Interestingly, epigenetic and gene expression studies unveiled the existence of different subtypes of SCLC based on their relative expression of the neuroendocrine lineage transcription factors ASCL1 (achaete-scute homologue 1) and NeuroD1 (neurogenic differentiation factor D1) and other transcription factors [26]. The existence of four subtypes has been confirmed by different studies [27][28][29] and a nomenclature for SCLC molecular subtypes has recently been proposed [30]. SCLC-A is the major group and displays high expression of ASCL1 (and its transcriptional target DLL3) and low expression of NeuroD1 markers. SCLC-N is the second most frequent subtype, and is characterized by high NeuroD1 and low (albeit variable) ASCL1 gene expression. SCLC-Y and SCLC-P represent smaller groups and comprise ASCL1/NeuroD1/INSM1 (insulinoma-associated protein 1, a neuroendocrine differentiation-promoting transcription factor) low tumors. These two subgroups, referred to as non-neuroendocrine, double negative or SQ-P (resembling primitive squamous tumors) by different studies [30] are characterized by the expression of the transcriptional co-activator YAP1 and the transcriptional factor POU2F3, respectively. Thus, it has become apparent that although loss of function of TP53 and/or RB1 is required for SCLC development, the existence of different subtypes is governed by gene expression programs under the control of specific transcriptional regulators.
Genomic profiling of LCNEC tumors has shown frequent mutations in TP53 and RB1, although the prevalence of RB1 mutations is lower than in SCLC. Recently, the first comprehensive characterization of LCNEC, combining sets of gene mutations with patterns of gene expression and comparing them to those in SCLC, highlighted differences and commonalities between the two types of high-grade neuroendocrine tumors. This study identified two molecular subgroups within LCNEC, termed type I and type II. Type I LCNEC displayed frequent mutations in TP53 and two other genes, STK11 (also known as LKB1, which encodes for a serine/threonine kinase that regulates cell polarity) and KEAP1 (involved in oxidative stress response), but not in RB1. Type II LCNEC bore frequent mutations in TP53 and RB1, and from that standpoint one would expect it to be the most similar to SCLC. In fact, two major LCNEC subsets had previously been identified based on their profile of altered genes, one considered SCLC-like, with frequent TP53/RB1 mutations, and the other nonSCLC-like, with frequent TP53/STK11/KEAP1/KRAS mutations. However, transcriptional profiles of type I and II LCNEC indicated the opposite. Type I LCNEC showed high levels of expression of ASCL1 and DLL3, similar to SCLC-A (ASCL-high/NeuroD1-high) tumors, and low levels of NOTCH. Conversely, type II LCNEC had reduced expression of ASCL1, DLL3, and other neuroendocrine genes, and high expression of NOTCH and immune-related pathways.
In contrast to high-grade neuroendocrine carcinomas, well-differentiated neuroendocrine tumors TC and AC show low mutation rate and rare mutations in TP53 and RB1 genes. Pulmonary carcinoids display frequent alterations in chromatin-remodeling genes such as MEN1 (the most frequently mutated), ARID1A and components of the SWI/SNF complex [31]. Comprehensive multi-omic analysis identified different molecular subgroups within well-differentiated neuroendocrine tumors. Molecular group A (further divided into A1 and A2) consisted mainly of TC; group B, enriched in AC; and supra-carcinoids, with very few samples. While high-grade neuroendocrine tumors and well-differentiated carcinoids possibly represent different diseases and not just a range of tumors with common pathogeneses, these comprehensive studies reveal the existence of molecular links between both groups of lung neuroendocrine tumors. Thus, the A1 subgroup of pulmonary carcinoids showed high expression of ASCL1 and DLL3 genes, which is similar to SCLC-A and type I LCNEC. The supra-carcinoid subgroup displayed molecular characteristics similar to LCNEC, including high expression of immune checkpoint genes. In fact, one case of LCNEC clustered in this group. This observation has led to the suggestion that supra-carcinoids could represent the lung equivalent to grade-3 neuroendocrine tumors present in other organs; currently this grade is only considered for the high-grade lung neuroendocrine SCLC and LCNEC [32].
3. Modelling Neuroendocrine Lung Cancer with GEMMs
Mouse models of neuroendocrine lung cancer based on patient-derived mutations are used to determine the mechanisms by which the disease arises and progresses. Advances in genetics, sequencing, and transcriptomic studies have identified disease-associated genetic alterations in neuroendocrine tumors. To determine causality between genetics and disease, accurate models for molecular dissection are required.
GEMMs are essential tools to delineate the multistage pathogenesis of human tumors, behavior, development and extension, progression, and therapy response. GEMMs can be used to induce spontaneous tumor growth via conventional transgenic or conditional mechanisms in a native immunocompetent microenvironment. Transgenic mice are generated by microinjection of DNA into the pronucleus of zygotes or injection of embryonic stem cells into blastocysts, to produce the desired loss, gain of function mutations, or substitutions (knock out or knock in). The currently available tools allow us to constitutively or conditionally activate or inactivate genes in distinct cell types at any desired moment. So far, the most widely used strategy in modeling neuroendocrine lung cancer is based on the ability of cre recombinase to direct site-specific DNA recombination between pairs of loxP sites. The use of fluorescent reporters that mark the switched cells helps to track and isolate cancer cells as tumor growth progresses from initial lesions to metastasis. Bioluminescent reporters also enable us to conduct lineage tracing, monitoring and quantification of the tumor in situ [33][34][35]. A drawback of current mouse models that harbor multiple genetic modifications is the time investment required to breed mice to generate such complex genotypes. This costly process involves hundreds of animals and takes years. Recent advances, including CRISPR/Cas9, have been used in vivo to manipulate specific genes and specific populations of cells [36][37] thus allowing faster generation of GEMMs which develop SCLC. Hopefully, additional neuroendocrine lung cancer models will soon be available by means of these techniques.
Another point to consider is that human tumors usually harbor a high burden of point mutations caused by cigarette smoke exposure, while tumors in GEMMs develop mostly due to targeted gene rearrangements. At any rate, such models serve to distinguish passenger from driver mutations. Exposure to chemical carcinogens (including smoke exposure) was one of the main focuses of the early lung cancer studies using mouse models, that lead to the development of adenocarcinoma in the majority of cases [38][39]. However, neuroendocrine lung tumors are virtually never found in spontaneous or chemically induced lung cancer models. This could be explained at least in part because most of the carcinogens, for which the mechanism of action is known, induce the activation of oncogenes such as K-ras [40][41][42], while the combinations of Trp53 and Rb alterations, responsible for the development of high-grade neuroendocrine tumors, are almost never found. Nevertheless, when applied to Rb-family deficient GEMMs, chemical carcinogens induce exclusively neuroendocrine tumors.
Some lung tumors modelled in mice accurately mimic their human counterparts [33], and one possible explanation resides in the feasibility of a refined method to neatly access mouse lungs: intratracheal infection of adenovirus in adult lungs has proven to be a robust method for modelling lung cancer [43]. Another contributing factor is the availability of specific cell promoters that determine the cell-type specificity of the genetic modification. Specific cell promoters are active in the different lung cell types: the rat CGRP promoter was identified as a neuroendocrine cell-specific promoter [44]; the 3.7-kb fragment of the human SPC promoter activity is restricted to alveolar type 2 (AT2) cells [45]. Similarly, Aquaporin 5 (Aqp5) promoter activity is constrained to alveolar type I cells [46]; the mouse Scgb1a1 (Secretoglobin1a1, also known as CCSP, CC10, and CCA) promoter fragment mainly targets bronchiolar Clara cells [47]. Although not expressed exclusively in the respiratory system, Keratin 5 (K5) and Keratin 14 (K14) promoters have been used to target basal cells in the airway epithelium [48]. Similarly, a 1-kb fragment of the human FOXJ1 promoter directs reporter gene expression to all ciliated cells including those of the lung, oviducts, ependyma, and testis [49].
Cell of Origin of SCLC
The utilization of adenoviruses carrying different promoters to direct cre expression to specific epithelial cell types has been particularly successful in dissecting the cell of origin of SCLC. This type of data can only be inferred from animal models. As different promoters targeting specific populations in the lung epithelia are employed in the construction of distinct adeno-cre viruses, genetic recombination is achieved in a cell-type restricted manner. Thus, SCLC has been reported to arise mainly from neuroendocrine cells (using a CGRP promoter) [50][51], and recent work using a neuroendocrine specific driver (Ascl1creERT2) showed that SCLC tumors can arise from a small subpopulation of rare, differentiated neuroendocrine stem cells [52]. However, there is strong evidence supporting that SCLC tumors can arise from other cell types in the lung. It is of note that the use of viruses carrying the SPC promoter, which targets AT2 cells, and K5 and K14 promoters that target keratin K5 and keratin K14 expressing epithelial basal cells, respectively, render SCLC [53]. In contrast, Sutherland et al. [50] reported no tumor formation when using the CC10 promoter that targets Clara cells. Taken together, data gathered from animal models support the idea that non-neuroendocrine lung epithelial cells may also serve as the cell type of origin for SCLC, at least in specific genetic contexts [53]. This suggests high cellular plasticity and an interplay—at least in mice—between targeted cells and modified genes. The question of the extent to which this is the case in humans remains open. Otherwise, in spite of the available tools, the cell(s) of origin of the other neuroendocrine lung tumor types (TC, AC and LCNEC) remain unknown.
Specifically targeting genetic alterations to different cell types has not only served the purpose of identifying the cell of origin from which SCLC arises, but also has revealed the existence of different metastatic programs [54], an activating or suppressive role of FGFR1 [53], and determine that the same set of genetic alterations can generate different types of high-grade neuroendocrine tumor [55]. A thorough review of the cells of origin of lung cancer and its influence in inter- and intratumoral heterogeneity has recently been published [56].
4. Animal Models of Pulmonary Carcinoids
There is a notable paucity of biological material for the study of these rare tumors, which has impeded significant advances in patient treatment. The studies of tumor biology using pulmonary carcinoids have been hindered by a lack of in vitro and in vivo models representing the carcinoid phenotype and behavior. Hardly a handful of different typical carcinoid cell lines have been generated [57], and two of them (NCI-H720 and NCI-H727) give rise to tumors when orthotopically injected into mice [58]. NCI-H727 [59] is the most used cell line for the study of typical carcinoma. This cell line has been used to test several compounds or molecular imaging techniques in different studies [60][61][62][63][64][65]. However, the characteristics of the primary tumors originating from this cell line are not consistent with the biology of a typical carcinoid, thus hampering the understanding of the molecular mechanisms underlying this tumor type. Chemically induced GEMMs for TC and AC exist: a triple knockout mutant model in which all the three Rb family members are ablated develops TC after DHPN (N-bis(2-hydroxypropyl) nitrosamine, a potent mutagen and a wide-spectrum carcinogen in rodents) administration and AC after urethane (an inducer of K-ras activation) treatment.
Thus, as mentioned above, and although the worldwide incidence of pulmonary carcinoids is increasing, modeling these tumor types in the mouse is still in its infancy [60] and implies an unmet need for studying the biology and novel intervention strategies for these tumors. Given the mutational pattern of MEN1, ARID1, EIF1AX, the SW1/SNF complex, ATM, PSIP1, and ROBO1 reported in human low-grade neuroendocrine tumors, it would be worthwhile to generate lung preclinical models based on these genetic changes aiming to recapitulate the human types and serve for the development and testing of new therapeutic approaches, particularly for the pulmonary carcinoids that metastasize and subsequently worsen their prognosis. In fact, several MEN1 conditional and knockout models have been generated that mimic neuroendocrine disorders and tumors in other organs different from the lung such as pancreas, thyroid, adrenal, and pituitary glands [60]. Combining approaches that target specific cell types in the lung with the introduction of the appropriate genetic changes found in human pulmonary carcinoids, the scientific community should be able to generate TC and AC mouse models resembling their human counterparts. These models will undoubtedly allow progress in the understanding of these understudied diseases.
5. Animal Models of LCNEC
A defined GEMM model of LCNEC based on the ablation of four tumor suppressors (Rb, Rbl1, Pten, Trp53) in a wide variety of lung epithelial cell types (using the CMV promoter) has recently been described. It is interesting to note that, in this quadruple mutant mouse, differential development of LCNEC or SCLC resides in the targeted cell initiating tumor progression. In this model, restricted cell type targeting to basal cells gives rise predominantly to SCLC, but a spectrum of neuroendocrine tumors is observed suggesting a close relationship between both tumoral types. Along these lines, in 2015, Gazdar et al. [66] published a thorough histopathological examination of relevant mouse models of SCLC. They found that most of the models evaluated depict the whole spectrum of high-grade neuroendocrine lung tumors, and in some stages of development the LCNEC component was predominant over the SCLC component. They also reported both components to be often intimately intermixed, suggesting high plasticity and transition from one to the other [66]. Akeno et al. also reported the formation of LCNEC along with SCLC in a Trp53 mutant and deficient Rb1 mouse [67]. However, the quadruple Rb, Rbl1, Pten, Trp53 mutant mice represent the first and only mouse model for LCNEC reported so far.
6. Animal Models of SCLC
In sharp contrast with the other types of pulmonary neuroendocrine tumors, a number of GEMMs for SCLC have been successfully reported. These tumors faithfully reproduce the histopathological features of the human condition (and the neuroendocrine nature of the tumor).
Based on the fact that p53 and RB1 are almost always inactivated in patients with SCLC, Meuwissen et al. [68] generated the first mouse model in which conditional deletion of Rb1 and Trp53 was accomplished by intratracheal adenoviral delivery of cre using the ubiquitous CMV promoter. The sporadic inactivation of both genes in a wide variety of lung epithelial cells renders (after long latency periods) spontaneous tumors that closely mimic the histopathology and metastatic behavior of human patients. This basic model was subsequently followed by variant versions when additional genetic modifications found or suspected in human SCLC tumors were introduced: inactivation of Rbl2, another member or the Rb family, resulted in accelerated SCLC tumor development [69]. This triple knock out Rb1/Trp53/Rbl2 maintains the histologic features and metastatic pattern of the disease. Loss of Pten, a gene frequently mutated in human SCLC, accelerated tumor progression as described by two groups: McFadden et al. reported SCLC tumor development when using the neuroendocrine-specific CGRP promoter; meanwhile, Cui et al. [70] observed a mixture of SCLC/LCNEC/ADC when using the CMV promoter. Combined deletion of two members of the retinoblastoma family, Rb1 and Rbl1, together with Pten and Trp53, resulted in a dramatic acceleration of SCLC tumor progression when epithelial basal cells were targeted by using the keratin K5 promoter .
Overexpression of Mycl, a transcription factor member of a family of oncogenes which was found amplified in ~9% of human SCLC together with the targeted deletion of Rb1 and Trp53 showed an earlier onset of SCLC and shortened latency of tumor development. In this model, metastasis is rarely seen [71][72]. However, a mutant MycT58A dramatically accelerates tumorigenesis and metastasis in the Rb1/Trp53 null SCLC promoting a variant phenotype [73]. These “myc models” also evidence the SCLC tumor heterogeneity. Similarly, overexpression of Nfib together with biallelic inactivation of Rb1 and Trp53 promoted earlier onset of SCLC and enhanced metastasis [74][75]. These triple or quadruple mutant models reduce the long latency periods of the first reported Rb1/Trp53 deficient mice, as they introduce the additional secondary genetic changes necessary for the tumor development. The role of Notch as a tumor suppressor in SCLC was established, as in the Rb1/Trp53/Rbl2 background there was a significant reduction of the number of tumors that developed when an activated form of NOTCH1 and NOTCH2 was overexpressed. Later on, subsequent models showed that Notch exerts its suppressor activities through different mechanisms.
These models have been invaluable tools to understand the biology of SCLC, for its preclinical intervention and translational research. Contrary to the other neuroendocrine lung tumor types, they make of SCLC one of the best represented diseases by GEMMs for the study of human cancer (Table 1). Gazdar et al. also remark on the importance of these GEMMs in providing the opportunity to study the early stages and the multistage pathogenesis of the disease, as SCLCs are seldom resected, thus hindering the opportunity of observing initial or preinvasive lesions in humans. These GEMMSs also reproduce the hepatic, mediastinal, and lymph node invasion seen in humans.
Table 1. Lung neuroendocrine cancer models obtained by intratracheal adenoviral cre delivery.
|
Tumor type |
|
Genotype |
Promoter |
Major Phenotype |
Ref. |
Comments |
|
TC |
|
Rb1/Rbl1/Rbl2 |
CMV |
TC |
[21] |
DHPN induction |
|
AC |
|
Rb1/Rbl1/Rbl2 |
CMV |
AC |
[21] |
Urethane induction |
|
LCNEC |
|
Rb1/Rbl1/Trp53/Pten |
CMV |
LCNEC |
[22] |
First LCNEC mouse model described |
|
SCLC |
|
Rb1/Trp53 |
CMV |
SCLC |
[68] |
First SCLC mouse model described |
|
|
|
Rb1/Trp53 |
CGRP, SPC |
SCLC |
[50] |
Cells of origin of SCLC |
|
|
Backbone Rb1/Trp53 AND |
|
|
|
|
|
|
|
|
Rbl2 |
CMV |
SCLC |
[69] |
Accelerated tumor development |
|
|
|
Rbl2 |
CGRP |
SCLC |
[54] |
Different metastatic program |
|
|
|
Pten |
CMV |
SCLC/LCNEC/ ADC |
[70] |
Accelerated tumor development |
|
|
|
Pten |
CGRP |
SCLC |
[76] |
Accelerated tumor development |
|
|
|
Pten/Rbl1 |
K5 |
SCLC |
[22] |
Basal cell of origin of SCLC |
|
|
|
Mycl |
CMV |
SCLC |
[71] |
Accelerated tumor development |
|
|
|
Mycl |
CMV |
SCLC |
[72] |
Heterogeneity and differential sensitivity to chemotherapy |
|
|
|
Myc T58A |
CGRP |
SCLC |
[73] |
SCLC-N subtype |
|
|
|
Myc T58A |
CGRP |
SCLC |
[77] |
Multiple subtypes present in a tumor |
|
|
|
Nfib |
CMV |
SCLC |
[75] |
Short latency and enhanced metastases |
|
|
|
Rbl2/Notch2 |
CMV |
SCLC |
[17] |
Role of Notch as tumor suppressor in SCLC |
|
|
|
Rbl2/NeuroD1 |
CMV |
SCLC |
[27] |
SCLC-A subtype |
|
|
|
FgFr1 |
K14 |
SCLC/ rare ADC |
[53] |
Context dependent effect of Fgfr1 |
TC: typical carcinoma; AT: atypical carcinoma; LCNEC: large cell neuroendocrine carcinoma; SCLC: small cell carcinoma; ADC: adenocarcinoma.
6.1. A Recent Classification of Subtypes of SCLC
Until recently, SCLC were considered a relatively homogenous lung cancer type. To date the WHO classification recognizes only one form of SCLC (in current clinical practice). This has led to similar chemotherapy treatment for patients in the last decades [78]. However, and as stated above, recent studies have reported a considerable amount of heterogeneity among SCLCs, mostly based on the studies that identify subtypes based on the expression of lineage-specific transcription factors. They also take into consideration expression of neuroendocrine markers [27], growth properties in cultures [79] and amplification or expression of MYC [73]. Animal models have a role in defining these subtypes by complementing data from cell lines and human tumors. The role of Ascl1 (and NeuroD1) in developing tumors of the Ascl1high (NeuroD1low) subtype was first reported in a mouse model generated by adding the inactivation of Ascl1 or Neuro D1 to the Rb1/Trp53/Rbl2 mouse. Interestingly, while loss Ascl1 impedes appearance of tumors, inactivation of NeuroD1 did not affect the development of SCLC [27]. The data further suggest that the preexisting SCLC models belong to the SCLC-A/Ascl1 high subtype. The mouse model representative of the NeuroD1 high is depicted in invasive tumors based on the activation of Myc and stabilization of Myc protein in addition to the loss of Rb1 and Trp53 (RPM mice) [73]. So far, no mouse models resulting in tumors representative of the SCLC-P or SCLC-Y groups have been generated. Presumably, future models will identify specific genetic events responsible for the development of these different tumor subtypes. However, a recent study using the RPM mouse model supports the conclusion that MYC drives (from a neuroendocrine cell of origin) the SCLC-A to SCLC-N to SCLC-Y subtypes in a temporal evolution. These data suggest that SCLC harbors cells in transcriptionally dynamic states of progression, and further depict the high intratumoral heterogeneity and plasticity of this disease [77]. This aspect of SCLC makes its subtyping even more complicated.
Considering that different subtypes have different therapeutic vulnerabilities, these recent models related to subtyping in classification help to clarify the identification of biomarkers, development, and application of targeted therapies, and more personalized treatment options, another unmet need in SCLC.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13010014
References
- Derks, J.L.; Leblay, N.; Lantuejoul, S.; Dingemans, A.C.; Speel, E.M.; Fernandez-Cuesta, L. New Insights into the Molecular Characteristics of Pulmonary Carcinoids and Large Cell Neuroendocrine Carcinomas, and the Impact on Their Clinical Management. J. Thorac. Oncol. 2018, 13, 752–766, doi:10.1016/j.jtho.2018.02.002.
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072, doi:10.1200/JCO.2007.15.4377.
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242, doi:10.1097/JTO.0000000000000663.
- Rindi, G.; Klimstra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; de Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018, 31, 1770–1786, doi:10.1038/s41379-018-0110-y.
- Pelosi, G.; Sonzogni, A.; Harari, S.; Albini, A.; Bresaola, E.; Marchio, C.; Massa, F.; Righi, L.; Gatti, G.; Papanikolaou, N.; et al. Classification of pulmonary neuroendocrine tumors: New insights. Transl. Lung Cancer Res. 2017, 6, 513–529, doi:10.21037/tlcr.2017.09.04.
- Fujino, K.; Motooka, Y.; Hassan, W.A.; Ali Abdalla, M.O.; Sato, Y.; Kudoh, S.; Hasegawa, K.; Niimori-Kita, K.; Kobayashi, H.; Kubota, I.; et al. Insulinoma-Associated Protein 1 Is a Crucial Regulator of Neuroendocrine Differentiation in Lung Cancer. Am. J. Pathol. 2015, 185, 3164–3177, doi:10.1016/j.ajpath.2015.08.018.
- Jia, S.; Wildner, H.; Birchmeier, C. Insm1 controls the differentiation of pulmonary neuroendocrine cells by repressing Hes1. Dev. Biol. 2015, 408, 90–98, doi:10.1016/j.ydbio.2015.10.009.
- Asamura, H.; Kameya, T.; Matsuno, Y.; Noguchi, M.; Tada, H.; Ishikawa, Y.; Yokose, T.; Jiang, S.X.; Inoue, T.; Nakagawa, K.; et al. Neuroendocrine neoplasms of the lung: A prognostic spectrum. J. Clin. Oncol. 2006, 24, 70–76, doi:10.1200/JCO.2005.04.1202.
- Caplin, M.E.; Baudin, E.; Ferolla, P.; Filosso, P.; Garcia-Yuste, M.; Lim, E.; Oberg, K.; Pelosi, G.; Perren, A.; Rossi, R.E.; et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoids. Ann. Oncol. 2015, 26, 1604–1620, doi:10.1093/annonc/mdv041.
- Naraev, B.G.; Ramirez, R.A.; Kendi, A.T.; Halfdanarson, T.R. Peptide Receptor Radionuclide Therapy for Patients With Advanced Lung Carcinoids. Clin. Lung Cancer 2019, 20, e376–e392, doi:10.1016/j.cllc.2019.02.007.
- Zimmerman, S.; Das, A.; Wang, S.; Julian, R.; Gandhi, L.; Wolf, J. 2017–2018 Scientific Advances in Thoracic Oncology: Small Cell Lung Cancer. J. Thorac. Oncol. 2019, 14, 768–783, doi:10.1016/j.jtho.2019.01.022.
- Rossi, G.; Cavazza, A.; Marchioni, A.; Longo, L.; Migaldi, M.; Sartori, G.; Bigiani, N.; Schirosi, L.; Casali, C.; Morandi, U.; et al. Role of chemotherapy and the receptor tyrosine kinases KIT, PDGFRalpha, PDGFRbeta, and Met in large-cell neuroendocrine carcinoma of the lung. J. Clin. Oncol. 2005, 23, 8774–8785, doi:10.1200/JCO.2005.02.8233.
- Hendifar, A.E.; Marchevsky, A.M.; Tuli, R. Neuroendocrine Tumors of the Lung: Current Challenges and Advances in the Diagnosis and Management of Well-Differentiated Disease. J. Thorac. Oncol. 2017, 12, 425–436, doi:10.1016/j.jtho.2016.11.2222.
- Derks, J.L.; Hendriks, L.E.; Buikhuisen, W.A.; Groen, H.J.; Thunnissen, E.; van Suylen, R.J.; Houben, R.; Damhuis, R.A.; Speel, E.J.; Dingemans, A.M. Clinical features of large cell neuroendocrine carcinoma: A population-based overview. Eur. Respir. J. 2016, 47, 615–624, doi:10.1183/13993003.00618-2015.
- Govindan, R.; Page, N.; Morgensztern, D.; Read, W.; Tierney, R.; Vlahiotis, A.; Spitznagel, E.L.; Piccirillo, J. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: Analysis of the surveillance, epidemiologic, and end results database. J. Clin. Oncol. 2006, 24, 4539–4544, doi:10.1200/JCO.2005.04.4859.
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110, doi:10.1038/ng.2396.
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53, doi:10.1038/nature14664.
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116, doi:10.1038/ng.2405.
- Fernandez-Cuesta, L.; Peifer, M.; Lu, X.; Sun, R.; Ozretic, L.; Seidal, D.; Zander, T.; Leenders, F.; George, J.; Muller, C.; et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat. Commun. 2014, 5, 3518, doi:10.1038/ncomms4518.
- Alcala, N.; Leblay, N.; Gabriel, A.A.G.; Mangiante, L.; Hervas, D.; Giffon, T.; Sertier, A.S.; Ferrari, A.; Derks, J.; Ghantous, A.; et al. Integrative and comparative genomic analyses identify clinically relevant pulmonary carcinoid groups and unveil the supra-carcinoids. Nat. Commun. 2019, 10, 3407, doi:10.1038/s41467-019-11276-9.
- Lazaro, S.; Perez-Crespo, M.; Enguita, A.B.; Hernandez, P.; Martinez-Palacio, J.; Oteo, M.; Sage, J.; Paramio, J.M.; Santos, M. Ablating all three retinoblastoma family members in mouse lung leads to neuroendocrine tumor formation. Oncotarget 2017, 8, 4373–4386, doi:10.18632/oncotarget.13875.
- Lazaro, S.; Perez-Crespo, M.; Lorz, C.; Bernardini, A.; Oteo, M.; Enguita, A.B.; Romero, E.; Hernandez, P.; Tomas, L.; Morcillo, M.A.; et al. Differential development of large-cell neuroendocrine or small-cell lung carcinoma upon inactivation of 4 tumor suppressor genes. Proc. Natl. Acad. Sci. USA 2019, 116, 22300–22306, doi:10.1073/pnas.1821745116.
- George, J.; Walter, V.; Peifer, M.; Alexandrov, L.B.; Seidel, D.; Leenders, F.; Maas, L.; Muller, C.; Dahmen, I.; Delhomme, T.M.; et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat. Commun. 2018, 9, 1048, doi:10.1038/s41467-018-03099-x.
- Miyoshi, T.; Umemura, S.; Matsumura, Y.; Mimaki, S.; Tada, S.; Makinoshima, H.; Ishii, G.; Udagawa, H.; Matsumoto, S.; Yoh, K.; et al. Genomic Profiling of Large-Cell Neuroendocrine Carcinoma of the Lung. Clin. Cancer Res. 2017, 23, 757–765, doi:10.1158/1078-0432.CCR-16-0355.
- Rekhtman, N.; Pietanza, M.C.; Hellmann, M.D.; Naidoo, J.; Arora, A.; Won, H.; Halpenny, D.F.; Wang, H.; Tian, S.K.; Litvak, A.M.; et al. Next-Generation Sequencing of Pulmonary Large Cell Neuroendocrine Carcinoma Reveals Small Cell Carcinoma-like and Non-Small Cell Carcinoma-like Subsets. Clin. Cancer Res. 2016, 22, 3618–3629, doi:10.1158/1078-0432.CCR-15-2946.
- Poirier, J.T.; Gardner, E.E.; Connis, N.; Moreira, A.L.; de Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015, 34, 5869–5878, doi:10.1038/onc.2015.38.
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 2016, 16, 1259–1272, doi:10.1016/j.celrep.2016.06.081.
- McColl, K.; Wildey, G.; Sakre, N.; Lipka, M.B.; Behtaj, M.; Kresak, A.; Chen, Y.; Yang, M.; Velcheti, V.; Fu, P.; et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 2017, 8, 73745–73756, doi:10.18632/oncotarget.20572.
- Huang, Y.H.; Klingbeil, O.; He, X.Y.; Wu, X.S.; Arun, G.; Lu, B.; Somerville, T.D.D.; Milazzo, J.P.; Wilkinson, J.E.; Demerdash, O.E.; et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 2018, 32, 915–928, doi:10.1101/gad.314815.118.
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. 2019, 19, 289–297, doi:10.1038/s41568-019-0133-9.
- Simbolo, M.; Mafficini, A.; Sikora, K.O.; Fassan, M.; Barbi, S.; Corbo, V.; Mastracci, L.; Rusev, B.; Grillo, F.; Vicentini, C.; et al. Lung neuroendocrine tumours: Deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J. Pathol. 2017, 241, 488–500, doi:10.1002/path.4853.
- Fernandez-Cuesta, L.; Foll, M. Molecular studies of lung neuroendocrine neoplasms uncover new concepts and entities. Transl. Lung Cancer Res. 2019, 8, S430–S434, doi:10.21037/tlcr.2019.11.08.
- Kwon, M.C.; Berns, A. Mouse models for lung cancer. Mol. Oncol. 2013, 7, 165–177, doi:10.1016/j.molonc.2013.02.010.
- Semenova, E.A.; Nagel, R.; Berns, A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015, 29, 1447–1462, doi:10.1101/gad.263145.115.
- Shue, Y.T.; Lim, J.S.; Sage, J. Tumor heterogeneity in small cell lung cancer defined and investigated in pre-clinical mouse models. Transl. Lung Cancer Res. 2018, 7, 21–31, doi:10.21037/tlcr.2018.01.15.
- Oser, M.G.; Sabet, A.H.; Gao, W.; Chakraborty, A.A.; Schinzel, A.C.; Jennings, R.B.; Fonseca, R.; Bonal, D.M.; Booker, M.A.; Flaifel, A.; et al. The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes Dev. 2019, 33, 1718–1738, doi:10.1101/gad.328336.119.
- Ng, S.R.; Rideout, W.M., 3rd; Akama-Garren, E.H.; Bhutkar, A.; Mercer, K.L.; Schenkel, J.M.; Bronson, R.T.; Jacks, T. CRISPR-mediated modeling and functional validation of candidate tumor suppressor genes in small cell lung cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 513–521, doi:10.1073/pnas.1821893117.
- Gazdar, A.F.; Hirsch, F.R.; Minna, J.D. From Mice to Men and Back: An Assessment of Preclinical Model Systems for the Study of Lung Cancers. J. Thorac. Oncol. 2016, 11, 287–299, doi:10.1016/j.jtho.2015.10.009.
- de Seranno, S.; Meuwissen, R. Progress and applications of mouse models for human lung cancer. Eur. Respir. J. 2010, 35, 426–443, doi:10.1183/09031936.00124709.
- Westcott, P.M.; Halliwill, K.D.; To, M.D.; Rashid, M.; Rust, A.G.; Keane, T.M.; Delrosario, R.; Jen, K.Y.; Gurley, K.E.; Kemp, C.J.; et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2015, 517, 489–492, doi:10.1038/nature13898.
- You, M.; Candrian, U.; Maronpot, R.R.; Stoner, G.D.; Anderson, M.W. Activation of the Ki-ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain A mouse. Proc. Natl. Acad. Sci. USA 1989, 86, 3070–3074.
- Nuzum, E.O.; Malkinson, A.M.; Beer, D.G. Specific Ki-ras codon 61 mutations may determine the development of urethan-induced mouse lung adenomas or adenocarcinomas. Mol. Carcinog. 1990, 3, 287–295.
- DuPage, M.; Dooley, A.L.; Jacks, T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 2009, 4, 1064–1072.
- Johnston, D.; Hatzis, D.; Sunday, M.E. Expression of v-Ha-ras driven by the calcitonin/calcitonin gene-related peptide promoter: A novel transgenic murine model for medullary thyroid carcinoma. Oncogene 1998, 16, 167–177, doi:10.1038/sj.onc.1201478.
- Wert, S.E.; Glasser, S.W.; Korfhagen, T.R.; Whitsett, J.A. Transcriptional elements from the human SP-C gene direct expression in the primordial respiratory epithelium of transgenic mice. Dev. Biol. 1993, 156, 426–443, doi:10.1006/dbio.1993.1090.
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605, doi:10.1002/dvg.20335.
- Li, H.; Cho, S.N.; Evans, C.M.; Dickey, B.F.; Jeong, J.W.; DeMayo, F.J. Cre-mediated recombination in mouse Clara cells. Genesis 2008, 46, 300–307, doi:10.1002/dvg.20396.
- Ferone, G.; Song, J.Y.; Sutherland, K.D.; Bhaskaran, R.; Monkhorst, K.; Lambooij, J.P.; Proost, N.; Gargiulo, G.; Berns, A. SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell 2016, 30, 519–532, doi:10.1016/j.ccell.2016.09.001.
- Ostrowski, L.E.; Hutchins, J.R.; Zakel, K.; O'Neal, W.K. Targeting expression of a transgene to the airway surface epithelium using a ciliated cell-specific promoter. Mol. Ther. 2003, 8, 637–645, doi:10.1016/s1525-0016(03)00221-1.
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.Y.; Berns, A. Cell of origin of small cell lung cancer: Inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011, 19, 754–764.
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell 2014, 156, 1298–1311, doi:10.1016/j.cell.2014.02.031.
- Ouadah, Y.; Rojas, E.R.; Riordan, D.P.; Capostagno, S.; Kuo, C.S.; Krasnow, M.A. Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell 2019, 179, 403–416.e23, doi:10.1016/j.cell.2019.09.010.
- Ferone, G.; Song, J.Y.; Krijgsman, O.; van der Vliet, J.; Cozijnsen, M.; Semenova, E.A.; Adams, D.J.; Peeper, D.; Berns, A. FGFR1 Oncogenic Activation Reveals an Alternative Cell of Origin of SCLC in Rb1/p53 Mice. Cell Rep. 2020, 30, 3837–3850.e3, doi:10.1016/j.celrep.2020.02.052.
- Yang, D.; Denny, S.K.; Greenside, P.G.; Chaikovsky, A.C.; Brady, J.J.; Ouadah, Y.; Granja, J.M.; Jahchan, N.S.; Lim, J.S.; Kwok, S.; et al. Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov. 2018, 8, 1316–1331, doi:10.1158/2159-8290.CD-17-0987.
- Santos, M. New models of large-cell neuroendocrine carcinoma and small-cell lung carcinoma. Mol. Cell. Oncol. 2020, 10.1080/23723556.2019.1702413, 1702413, doi:10.1080/23723556.2019.1702413.
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of origin of lung cancers: Lessons from mouse studies. Genes Dev. 2020, 34, 1017–1032, doi:10.1101/gad.338228.120.
- Asiedu, M.K.; Thomas, C.F., Jr.; Tomaszek, S.C.; Peikert, T.; Sanyal, B.; Sutor, S.L.; Aubry, M.C.; Li, P.; Wigle, D.A. Generation and sequencing of pulmonary carcinoid tumor cell lines. J. Thorac. Oncol. 2014, 9, 1763–1771, doi:10.1097/JTO.0000000000000339.
- Bayat Mokhtari, R.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Sparaneo, A.; Muscarella, L.A.; Zhao, S.; Cheng, H.L.; Das, B.; Yeger, H. Human bronchial carcinoid tumor initiating cells are targeted by the combination of acetazolamide and sulforaphane. BMC Cancer 2019, 19, 864, doi:10.1186/s12885-019-6018-1.
- Phelps, R.M.; Johnson, B.E.; Ihde, D.C.; Gazdar, A.F.; Carbone, D.P.; McClintock, P.R.; Linnoila, R.I.; Matthews, M.J.; Bunn, P.A., Jr.; Carney, D.; et al. NCI-Navy Medical Oncology Branch cell line data base. J. Cell Biochem. Suppl. 1996, 24, 32–91, doi:10.1002/jcb.240630505.
- Gahete, M.D.; Jimenez-Vacas, J.M.; Alors-Perez, E.; Herrero-Aguayo, V.; Fuentes-Fayos, A.C.; Pedraza-Arevalo, S.; Castano, J.P.; Luque, R.M. Mouse models in endocrine tumors. J. Endocrinol. 2018, 10.1530/JOE-18-0571, doi:10.1530/JOE-18-0571.
- Johnbeck, C.B.; Munk Jensen, M.; Haagen Nielsen, C.; Fisker Hag, A.M.; Knigge, U.; Kjaer, A. 18F-FDG and 18F-FLT-PET imaging for monitoring everolimus effect on tumor-growth in neuroendocrine tumors: Studies in human tumor xenografts in mice. PLoS ONE 2014, 9, e91387, doi:10.1371/journal.pone.0091387.
- Oxboel, J.; Brandt-Larsen, M.; Schjoeth-Eskesen, C.; Myschetzky, R.; El-Ali, H.H.; Madsen, J.; Kjaer, A. Comparison of two new angiogenesis PET tracers 68Ga-NODAGA-E[c(RGDyK)]2 and (64)Cu-NODAGA-E[c(RGDyK)]2; in vivo imaging studies in human xenograft tumors. Nucl. Med. Biol. 2014, 41, 259–267, doi:10.1016/j.nucmedbio.2013.12.003.
- Severin, G.W.; Kristensen, L.K.; Nielsen, C.H.; Fonslet, J.; Jensen, A.I.; Frellsen, A.F.; Jensen, K.M.; Elema, D.R.; Maecke, H.; Kjaer, A.; et al. Neodymium-140 DOTA-LM3: Evaluation of an In Vivo Generator for PET with a Non-Internalizing Vector. Front. Med. 2017, 4, 98, doi:10.3389/fmed.2017.00098.
- Lee, M.J.; Miller, Z.; Park, J.E.; Bhattarai, D.; Lee, W.; Kim, K.B. H727 cells are inherently resistant to the proteasome inhibitor carfilzomib, yet require proteasome activity for cell survival and growth. Sci. Rep. 2019, 9, 4089, doi:10.1038/s41598-019-40635-1.
- Lundsten, S.; Spiegelberg, D.; Stenerlow, B.; Nestor, M. The HSP90 inhibitor onalespib potentiates 177LuDOTATATE therapy in neuroendocrine tumor cells. Int. J. Oncol. 2019, 55, 1287–1295, doi:10.3892/ijo.2019.4888.
- Gazdar, A.F.; Savage, T.K.; Johnson, J.E.; Berns, A.; Sage, J.; Linnoila, R.I.; MacPherson, D.; McFadden, D.G.; Farago, A.; Jacks, T.; et al. The comparative pathology of genetically engineered mouse models for neuroendocrine carcinomas of the lung. J. Thorac. Oncol. 2015, 10, 553–564, doi:10.1097/JTO.0000000000000459.
- Akeno, N.; Reece, A.L.; Callahan, M.; Miller, A.L.; Kim, R.G.; He, D.; Lane, A.; Moulton, J.S.; Wikenheiser-Brokamp, K.A. TRP53 Mutants Drive Neuroendocrine Lung Cancer Through Loss-of-Function Mechanisms with Gain-of-Function Effects on Chemotherapy Response. Mol. Cancer Ther. 2017, 16, 2913–2926, doi:10.1158/1535-7163.MCT-17-0353.
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189.
- Schaffer, B.E.; Park, K.S.; Yiu, G.; Conklin, J.F.; Lin, C.; Burkhart, D.L.; Karnezis, A.N.; Sweet-Cordero, E.A.; Sage, J. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010, 70, 3877–3883, doi:10.1158/0008-5472.CAN-09-4228.
- Cui, M.; Augert, A.; Rongione, M.; Conkrite, K.; Parazzoli, S.; Nikitin, A.Y.; Ingolia, N.; MacPherson, D. PTEN is a potent suppressor of small cell lung cancer. Mol. Cancer Res. 2014, 12, 654–659, doi:10.1158/1541-7786.MCR-13-0554.
- Huijbers, I.J.; Bin Ali, R.; Pritchard, C.; Cozijnsen, M.; Kwon, M.C.; Proost, N.; Song, J.Y.; de Vries, H.; Badhai, J.; Sutherland, K.; et al. Rapid target gene validation in complex cancer mouse models using re-derived embryonic stem cells. EMBO Mol. Med. 2014, 6, 212–225, doi:10.1002/emmm.201303297.
- Bottger, F.; Semenova, E.A.; Song, J.Y.; Ferone, G.; van der Vliet, J.; Cozijnsen, M.; Bhaskaran, R.; Bombardelli, L.; Piersma, S.R.; Pham, T.V.; et al. Tumor Heterogeneity Underlies Differential Cisplatin Sensitivity in Mouse Models of Small-Cell Lung Cancer. Cell Rep. 2019, 27, 3345–3358 e3344, doi:10.1016/j.celrep.2019.05.057.
- Mollaoglu, G.; Guthrie, M.R.; Bohm, S.; Bragelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285, doi:10.1016/j.ccell.2016.12.005.
- Denny, S.K.; Yang, D.; Chuang, C.H.; Brady, J.J.; Lim, J.S.; Gruner, B.M.; Chiou, S.H.; Schep, A.N.; Baral, J.; Hamard, C.; et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016, 166, 328–342, doi:10.1016/j.cell.2016.05.052.
- Semenova, E.A.; Kwon, M.C.; Monkhorst, K.; Song, J.Y.; Bhaskaran, R.; Krijgsman, O.; Kuilman, T.; Peters, D.; Buikhuisen, W.A.; Smit, E.F.; et al. Transcription Factor NFIB Is a Driver of Small Cell Lung Cancer Progression in Mice and Marks Metastatic Disease in Patients. Cell Rep. 2016, 16, 631–643, doi:10.1016/j.celrep.2016.06.020.
- McFadden, D.G.; Vernon, A.; Santiago, P.M.; Martinez-McFaline, R.; Bhutkar, A.; Crowley, D.M.; McMahon, M.; Sadow, P.M.; Jacks, T. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E1600–E1609, doi:10.1073/pnas.1404357111.
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78.e12, doi:10.1016/j.ccell.2020.05.001.