Mitochondrial microRNAs (mitomiRs) are endogenous small, single-stranded molecules of noncoding RNA (19–23 nucleotides) present in mitochondria that represent a new level of control of gene expression. These sequences can be either encoded in the nuclei - however the importing mechanism is still not fully established - or may be originated straight inside mitochondria, from mitochondrial genome-derived mRNA. Undeniably, mitomiRs typically act by regulating gene expression inside mitochondria at the post-transcriptional level with a significant role both in physiology and in pathology. Unveiling mitochondrial microRNAs biological function and their targets will propel the development of innovative therapeutic and diagnostic tools.
- Mitochondrial microRNAs,Tissue Regeneration
1. Introduction
MicroRNAs (miRNAs) are endogenous small, single-stranded molecules of noncoding RNA (19–23 nucleotides) that represent a new level of control of gene expression. miRNAs act at the post-transcriptional level to modulate protein-coding genes, either by blocking the translation of messenger RNA (mRNA) or by actively encouraging its degradation, and it is well reported that each miRNA can target multiple genes [1]. Given that miRNAs are now postulated as master regulators and fine tuners of gene expression, these molecules might enclose relevant diagnostic, prognostic and therapeutic applications.
miRNAs are currently documented as relevant players in almost every biological process, from cell proliferation to differentiation, apoptosis and organogenesis [1][2]. The finding and the study of miRNAs has noticeably changed the classical understanding of gene expression and our comprehension of the biogenesis and function of miRNAs has markedly grown in recent years. Some associations between miRNA deregulation and human disease have been reported in different medical fields [3]. The research in this new ‘arena’ has exposed the enormous potential of miRNAs as tools for diagnostics or therapeutics. More specifically, several studies have already explored the modulation of mitochondrial DNA genetics by miRNAs [4]. Mitochondria, besides chloroplasts in plants, are the only organelles that possess a separate genome, the so-called mitochondrial DNA (mtDNA). Mutations in this DNA molecule have been shown to be involved in an assortment of both physiological (e.g., heat production, reactive oxygen species (ROS) production, apoptosis, cellular differentiation and aging) and pathological traits, including neurodegenerative diseases, diabetes, metabolic syndrome and cancers [5][6]. Additionally, there is a plethora of crosstalk signals that allow the communication between the nuclear and mitochondrial genomes. This mechanism of regulation is vital for the activity of the whole cellular machinery, basically by modulating mitochondrial biogenesis and metabolism [7] through reciprocal mitochondrial-to-nucleus communication [8][9].
The most famous role of mitochondria is to generate ATP through oxidative phosphorylation fueled by a chain of four protein complexes, the electron transport chain (ETC). An impressive number of more than 1000 mitochondrial proteins have been discovered [10]. Mitochondrial proteins can have a distinct genetic origin. It is predicted that ~99% of these proteins are nuclear-encoded and are synthesized in the cytoplasmatic compartment, being further imported through mitochondrial membrane transporters. The lasting 1% of mitochondrial proteins are encoded by the mitochondrial genome and the mitochondrial ribosome (mitoribosome) is responsible for the translation of these mRNAs. Moreover, the mitochondrion has its own protein synthesis machinery. As a result, an appropriate regulation of mitochondrial protein synthesis is absolutely required to achieve and maintain normal mitochondrial function.
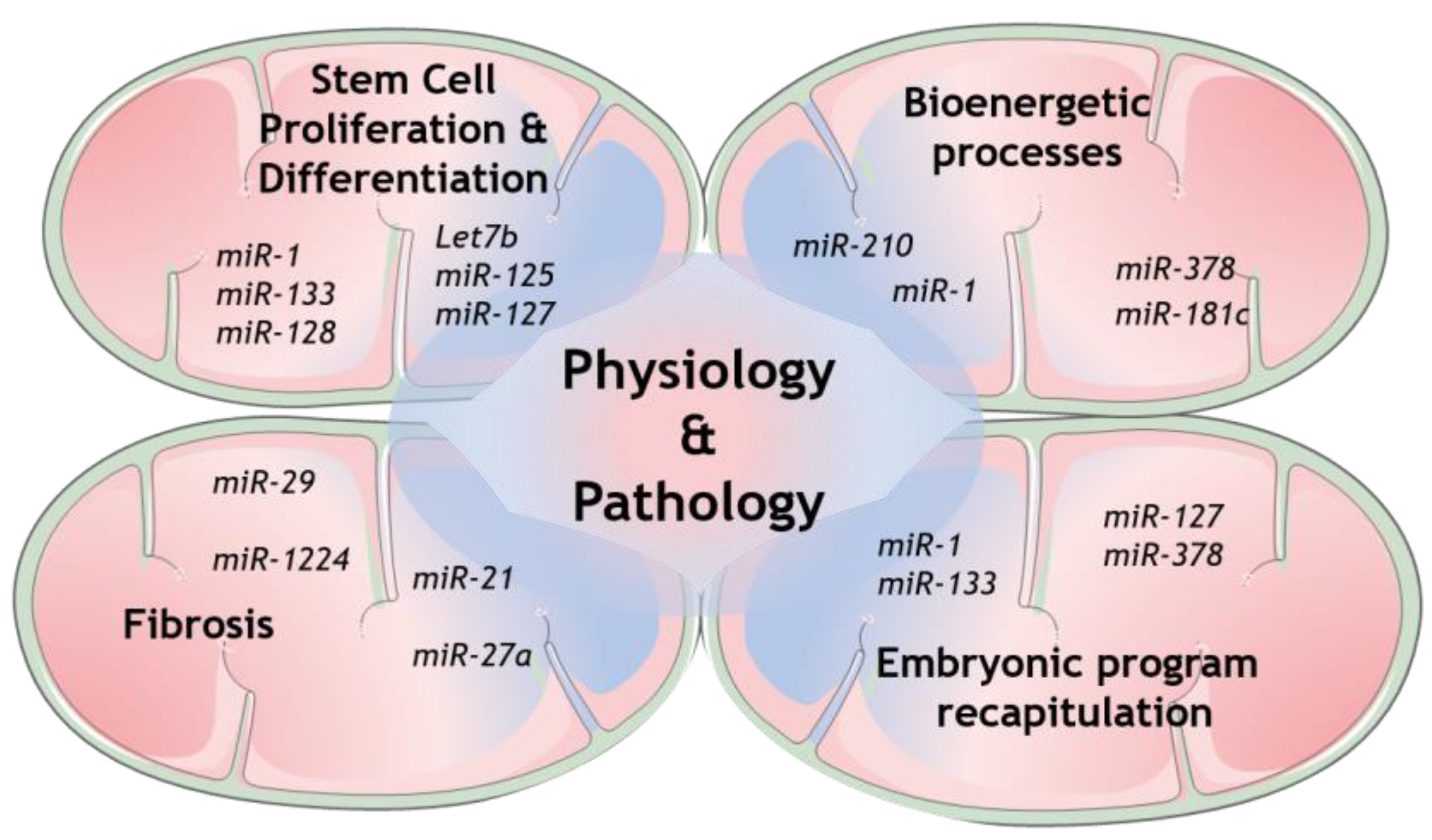
Regarding miRNAs in mitochondria, it is well-recognized nowadays that several cellular mechanisms involving mitochondria are regulated by many genetic players that originate from either nuclear- or mitochondrial-encoded small noncoding RNAs (sncRNAs). Growing evidence collected from whole genome and transcriptome sequencing highlight the role of distinct members of this class, from short interfering RNAs (siRNAs) to miRNAs and long noncoding RNAs (lncRNAs) [11]. Some of the mechanisms that have been shown to be modulated are the expression of mitochondrial proteins itself, as well as the more complex coordination of mitochondrial structure and dynamics with its function [12][13][14]. The mechanisms involved in mitochondrial miRNAs transport are now being increasingly exposed and therefore more and more light is being shed upon mitochondrial miRNAs’ targets to determine their role in this unmapped cellular context [11][15]. Indeed, several studies have already disclosed the presence of miRNAs in mitochondria [13]. However, the mechanism by which the nuclear-encoded miRNAs are imported into mitochondria is still not fully established (see Reference [15] for a follow-up). On the other hand, numerous mitomiRs may be originated straight inside mitochondria, from mitochondrial genome-derived mRNA. Undeniably, mitomiRs typically act by regulating gene expression inside mitochondria at the post-transcriptional level [16]. Further than that, some of the mitomiRs may also target nuclear-encoded mRNAs localized on the mitochondrial surface [11]. Taken all together, these discoveries clearly reveal the significant role of mitomiRs in regulating mitochondrial gene expression and mitochondrial functions in both physiology and pathology [17], as summarized in Figure 1.
2. Mitochondrial miRNAs—Potential Contribution for Regeneration
Tissue repair and regeneration depends on miRNA regulation as these small molecular silencers play a fundamental part in post-transcriptional gene silencing. Multiple key biological processes important for regeneration such as cell growth and proliferation, differentiation and apoptosis, as well as mitochondrial function, are tightly controlled by them [18][19]. Regulation of miRNA expression levels is crucial as small changes in basal conditions are sequentially propagated and amplified throughout different biological pathways, ultimately leading to changes in cell phenotypes [20]. Therefore, a plethora of aspects in tissue regeneration could potentially be controlled by the manipulation of miRNAs.
Currently, different evidence shows that both nuclear-encoded miRNAs imported to mitochondria and mitochondrial genome-derived miRNA, both defined as mitomiRs, may influence mitochondrial dynamics, a fundamental process for tissue homeostasis (Table 1) [21][22]. In fact, regulation of how and how much energy cells require strongly impacts stem cells’ fate, controlling the ability of stem cells to decide when to exit from their quiescent state.
Table 1. Summary of mitomiRs function and its known targets.
| miR | Target genes | Function | Reference |
|---|---|---|---|
| miR-1 | ↑ protein synthesis ↑ATP production |
[23] | |
| EIF4E, Mef2a, Gata4, HDAC6 | Regulation of cardiac hypertrophy | [24][25] | |
| FABP3 | Heart enlargement and hypertrophy | [26] | |
| Fibulin-2 | ↓ TGFβ signaling ↓ extracellular matrix remodeling |
[27] | |
| miR1/miR-133a | ↑ number of mitochondrial genesInfluence on mitochondria morphology | [28] | |
| ↑ cardiac stem cell differentiation | [29] | ||
| Let-7b | IGF-2 | ↓cell proliferation ↑ cell cycle arrest ↑ myofibroblast proliferation |
[30] |
| Hmga2 | ↑ cell senescence | [31] | |
| miR-127 | S1P3 | ↑ cell differentiation | [32] |
| ATP5B | Control of bioenergetic cell pattern | [33] | |
| miR-125b | IGF-2 | ↓ stem cell differentiation | [34] |
| miR-128 | Sp1 | ↓ stem cell differentiation | [35] |
| miR-181c | COX1 | Altered mitochondrial metabolism and ROS generation | [36][37] |
| miR-181a | ↑ cell senescence | [38] | |
| miR-338 | Modulate COX-IV and subunits of the ATP synthase complex | [39][40] | |
| miR-378 | ATP6 | ↓ ATP synthase activity | [41] |
| IGF receptor 1 | ↑ apoptosis ↓ signaling in Akt cascade ↑ ROS generation |
[42] | |
| miR-146-5p | ND1, ND2, ND4, ND5, ND6, ATP8, SOD3, Bcl-2 | ↑ ROS generation ↑ cell senescence |
[43][44][45] |
| miR-762 | ND2 | ↓ intracellular ATP levels ↑ increased ROS production ↓ mitochondrial complex I enzyme activity |
[46] |
| miR-19b, miR-20a, miR-17, miR-106 | Bcl-2 | ↑ permeability transition pore opening ↑ caspase-1 and 3 ↑ apoptosis |
[47][48] |
| miR-133 | type 1 angiotensin II receptor, Cdc42, Rho-A and Nelf-A/WHSC2 | ↓ cardiac remodeling | [49][50] |
| miR-212/132 | Foxo3 | ↑ cardiac remodeling | [51] |
| miR-23a | Foxo3 | ↑ cardiac remodeling | [52] |
| miR-29a-3p | NFATc4 | ↓ hypertrophic response | [53] |
| miR-101 | TGFβRI and c-Fos | ↓ Extracellular matrix production ↓ fibroblast proliferation |
[54][55] |
| miR-24 | furin | ↓ differentiation and migration of cardiac fibroblasts via TGFβ-smad2/3 | [56] |
| miR-29 family | Antifibrotic role | [56][57][58][59] | |
| miR-1224-3p | BECN1 | ↑ EMT ↓ gene expression of extracellular matrix-related genes |
[60] |
| miR-21 | Smad7 | Pro-fibrotic effect | [61] |
| Spry1 | Pro-fibrotic effect | [62] | |
| miR-27a | TGFb | Pro-fibrotic effect | [16][17] |
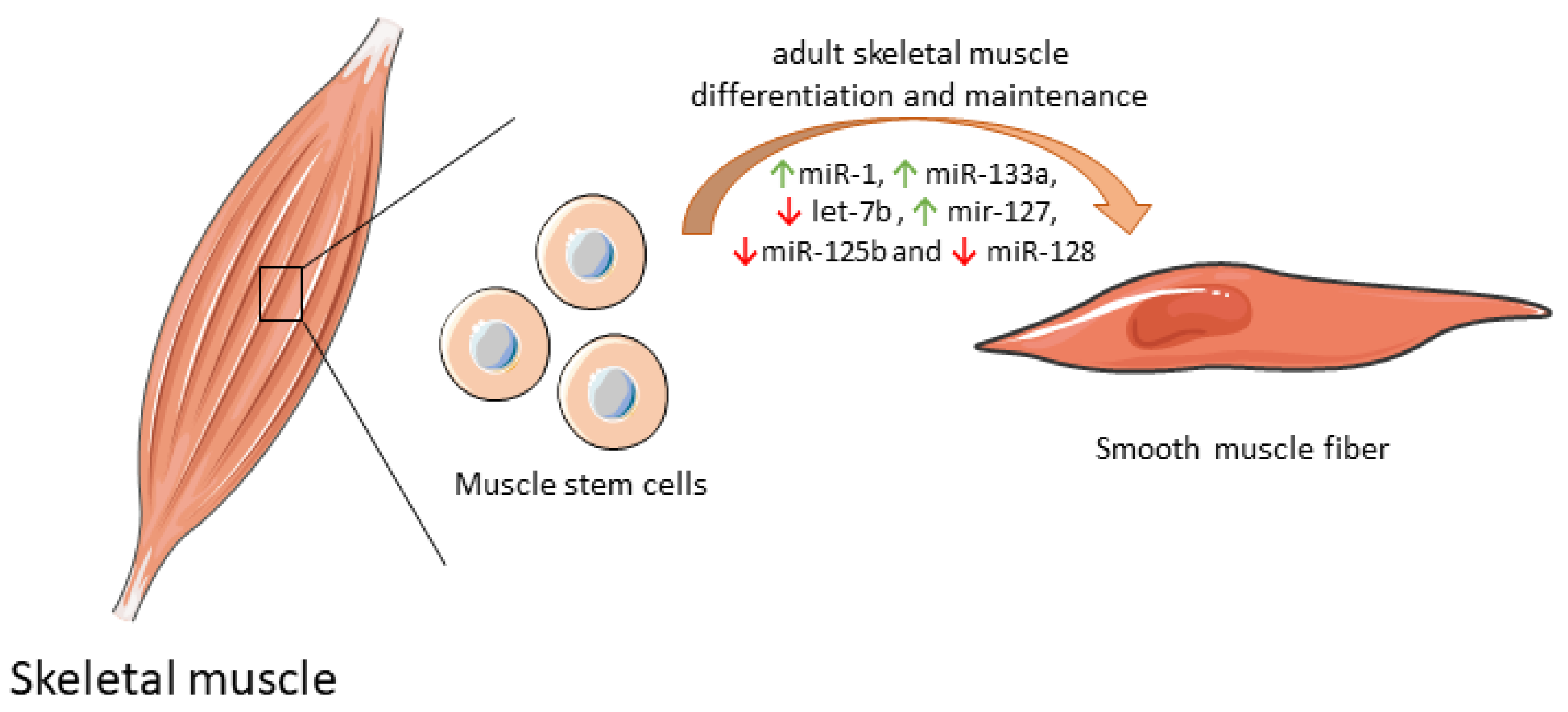
Some mitochondrial miRNAs were shown to modulate the differentiation of muscle stem cell into functional muscle cells by regulating mitochondria biogenesis (Table 1). By itself, miR-1 expression increases protein synthesis, and ATP production essential for optimal cell differentiation [35]. However, when miR-1 is simultaneously silenced with miR-133a in adult muscle stem cells in vitro, the expression of some mitochondrial genes was reduced, and atypical mitochondria were formed. Furthermore, a parallel in vivo experiment in a miR-1/133a double-knockout mouse model confirmed compromised muscle performance, as a consequence of mitochondrial dysfunction and impaired metabolic maturation [63]. let-7b, which has a dual role both on mitochondrial dynamics as well as on differentiation and maintenance of adult muscle cells [64], inhibited skeletal muscle growth by blocking cell proliferation and promoted cell cycle arrest and myofibroblast proliferation via the IGF-2 signaling pathway in vitro [36]. miR-127 was shown to mediate muscle stem cell differentiation through direct targeting sphingosine-1-phosphate receptor 3 (S1P3). Forced expression of miR-127 powered cell differentiation into skeletal cells both in vitro and in vivo. In a miR-127 transgenic mouse suffering Duchenne muscular dystrophy, administration of this biomolecule induced skeletal muscle regeneration and improved muscular dystrophy through stem cell differentiation [37]. Overexpression of other mitochondrial miRNAs, such as miR-125b and miR-128, was also correlated with inhibition of muscle stem cell differentiation and consequently impaired muscle regeneration [65]. Of note, miR-125b contributes to this inhibition through insulin-like growth factor 2 (IGF-2) targeting [39], while miR-128 suppresses specificity protein-1 (Sp1) [40]. In contrast, inhibition of miR-128 overexpresses Sp1 protein levels, abolishing proliferation and increasing differentiation [40]. The differential expression of mitomiRs that impact adult skeletal muscle cell differentiation and maintenance are depicted in Figure 2.
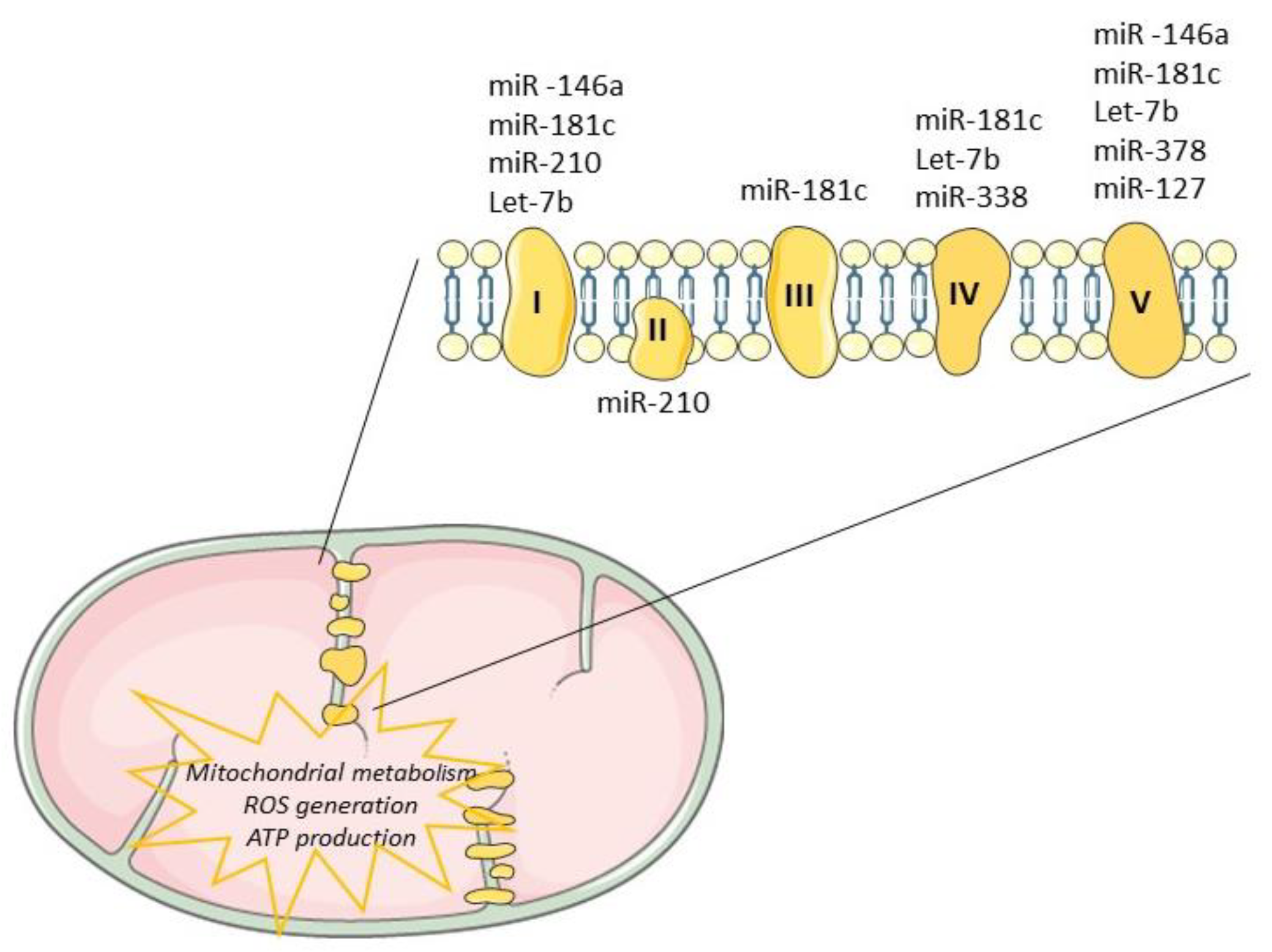
Tissue regeneration after injury is a complex and a highly metabolically demanding process [41]. Thus, regulation of the mitochondrial genome by mitomiRs may impact the expression of key components driving ATP synthesis, consequently influencing regenerative mechanisms (Table 1). Mitochondrial miR-181c, miR-1, miR-338 and miR-210 have been shown to target multiple proteins that modulate the mitochondrial electron transport chain [18][66], as depicted in Figure 3. The impact of miR-181c was demonstrated both in vitro [29] and in vivo [42] and mechanistically described by binding to 3’UTR mitochondrial cytochrome c oxidase subunit (COX)-1, which affects the function of mitochondrial electron transport chain complex IV. While forced overexpression of miR-181c altered mitochondrial metabolism and ROS generation, contributing to heart failure, its inhibition may be enough to balance mitochondrial bioenergetics, potentiating cardiac remodeling or at least controlling cardiac damages. Recently, Banavath et al. showed that the loss of miR-181c, through MICU1 upregulation, a specific promoter of Sp1, may protect the heart from injury [67]. Mitochondrial miR-338-5p in neural cell culture systems was demonstrated to modulate the expression of COX-IV and subunits of the ATP synthase complex [33][68]. When anti-miR-338 was transfected into axons, the metabolic oxygen consumption was increased by about 50% when compared with the nontargeting cells, potentiating cell survival. Furthermore, miR-378 was shown to have an important role in cardiac remodeling. This miRNA is highly expressed in diabetic cardiac mitochondria and it reduces ATP synthase activity by decreasing mitochondrial ATP6 expression. This effect was further tested in vitro by overexpression of mitomiR-378 in HL-1 cells [69]. Consistently, in vivo delivery of miR-378-3p antagomir preserved ATP6 protein levels which balance the bioenergetic deficits, contributing to an adequate cardiac pump function [69].
This entry is adapted from the peer-reviewed paper 10.3390/biology9120486
References
- Tétreault, N.; De Guire, V. MiRNAs: Their discovery, biogenesis and mechanism of action. Clin. Biochem. 2013, 46, 842–845.
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355.
- Condrat, C.E.; Thompson, D.C.; Barbu, M.G.; Bugnar, O.L.; Boboc, A.; Cretoiu, D.; Suciu, N.; Cretoiu, S.M.; Voinea, S.C. miRNAs as Biomarkers in Disease: Latest Findings Regarding Their Role in Diagnosis and Prognosis. Cells 2020, 9, 276.
- D’Aquila, P.; Bellizzi, D.; Passarino, G. Mitochondria in health, aging and diseases: The epigenetic perspective. Biogerontology 2015, 16, 569–585.
- Hudson, G.; Gomez-Duran, A.; Wilson, I.J.; Chinnery, P.F. Recent Mitochondrial DNA Mutations Increase the Risk of Developing Common Late-Onset Human Diseases. PLoS Genet. 2014, 10, e1004369.
- Dowling, D.K. Evolutionary perspectives on the links between mitochondrial genotype and disease phenotype. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1393–1403.
- Duarte, F.V.; Amorim, J.A.; Palmeira, C.M.; Rolo, A.P. Regulation of Mitochondrial Function and its Impact in Metabolic Stress. Curr. Med. Chem. 2015, 22, 2468–2479.
- Guha, M.; Avadhani, N.G. Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 2013, 13, 577–591.
- Whelan, S.P.; Zuckerbraun, B.S. Mitochondrial signaling: Forwards, backwards, and in between. Oxid. Med. Cell. Longev. 2013, 2013.
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284.
- Song, R.; Hu, X.Q.; Zhang, L. Mitochondrial MiRNA in Cardiovascular Function and Disease. Cells 2019, 8, 1475.
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Emerging Role of MitomiRs in the Pathophysiology of Human Disease. In Advances in Experimental Medicine and Biology; Santulli, G., Ed.; Springer: New York, NY, USA, 2015; Volume 888, pp. 123–154.
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014, 5, 865–886.
- Tomasetti, M.; Neuzil, J.; Dong, L. MicroRNAs as regulators of mitochondrial function: Role in cancer suppression. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1441–1453.
- Macgregor-Das, A.M.; Das, S. A microRNA’s journey to the center of the mitochondria. Am. J. Physiol. Circ. Physiol. 2018, 315, H206–H215.
- Fan, S.; Tian, T.; Chen, W.; Lv, X.; Lei, X.; Zhang, H.; Sun, S.; Cai, L.; Pan, G.; He, L.; et al. Mitochondrial miRNA Determines Chemoresistance by Reprogramming Metabolism and Regulating Mitochondrial Transcription. Cancer Res. 2019, 79, 1069–1084.
- Shinde, S.; Bhadra, U. A complex genome-MicroRNA interplay in human mitochondria. Biomed Res. Int. 2015, 2015.
- Sen, C.K.; Ghatak, S. miRNA control of tissue repair and regeneration. Am. J. Pathol. 2015, 185, 2629–2640.
- Borralho, P.M.; Rodrigues, C.M.P.; Steer, C.J. Mitochondrial MicroRNAs and Their Potential Role in Cell Function. Curr. Pathobiol. Rep. 2014, 2, 123–132.
- Sen, C.K. (Ed.) MicroRNA in Regenerative Medicine; Elsevier: Amsterdam, The Netherlands, 2015; ISBN 9780124055445.
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018, 430, 3922–3941.
- Sekar, D.; Johnson, J.; Biruntha, M.; Lakhmanan, G.; Gurunathan, D.; Ross, K. Biological and clinical relevance of microRNAs in mitochondrial diseases/dysfunctions. DNA Cell Biol. 2020, 39, 1379–1384.
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA Directly Enhances Mitochondrial Translation during Muscle Differentiation. Cell 2014, 158, 607.
- Jusic, A.; Devaux, Y. Mitochondrial noncoding RNA-regulatory network in cardiovascular disease. Basic Res. Cardiol. 2020, 115, 1–17.
- Chen, C.; Ponnusamy, M.; Liu, C.; Gao, J.; Wang, K.; Li, P. MicroRNA as a Therapeutic Target in Cardiac Remodeling. Biomed Res. Int. 2017, 2017, 1278436.
- Varrone, F.; Gargano, B.; Carullo, P.; Di Silvestre, D.; De Palma, A.; Grasso, L.; Di Somma, C.; Mauri, P.; Benazzi, L.; Franzone, A.; et al. The Circulating Level of FABP3 Is an Indirect Biomarker of MicroRNA-1. J. Am. Coll. Cardiol. 2013, 61, 88–95.
- Karakikes, I.; Chaanine, A.H.; Kang, S.; Mukete, B.N.; Jeong, D.; Zhang, S.; Hajjar, R.J.; Lebeche, D. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J. Am. Heart Assoc. 2013, 2, e000078.
- Wüst, S.; Dröse, S.; Heidler, J.; Wittig, I.; Klockner, I.; Franko, A.; Bonke, E.; Günther, S.; Gärtner, U.; Boettger, T.; et al. Metabolic Maturation during Muscle Stem Cell Differentiation Is Achieved by miR-1/133a-Mediated Inhibition of the Dlk1-Dio3 Mega Gene Cluster. Cell Metab. 2018, 27, 1026–1039.e6.
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121.
- Lin, S.; Luo, W.; Ye, Y.; Bekele, E.J.; Nie, Q.; Li, Y.; Zhang, X. Let-7b Regulates Myoblast Proliferation by Inhibiting IGF2BP3 Expression in Dwarf and Normal Chicken. Front. Physiol. 2017, 8, 477.
- Nishino, J.; Kim, I.; Chada, K.; Morrison, S.J. Hmga2 Promotes Neural Stem Cell Self-Renewal in Young but Not Old Mice by Reducing p16Ink4a and p19Arf Expression. Cell 2008, 135, 227–239.
- Zhai, L.; Wu, R.; Han, W.; Zhang, Y.; Zhu, D. miR-127 enhances myogenic cell differentiation by targeting S1PR3. Cell Death Dis. 2017, 8, e2707.
- Willers, I.M.; Martínez-Reyes, I.; Martínez-Diez, M.; Cuezva, J.M. miR-127-5p targets the 3′UTR of human β-F1-ATPase mRNA and inhibits its translation. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 838–848.
- Ge, Y.; Sun, Y.; Chen, J. IGF-II is regulated by microRNA-125b in skeletal myogenesis. J. Cell Biol. 2011, 192, 69–81.
- Dai, Y.; Zhang, W.R.; Wang, Y.M.; Liu, X.F.; Li, X.; Ding, X.B.; Guo, H. microRNA-128 regulates the proliferation and differentiation of bovine skeletal muscle satellite cells by repressing Sp1. Mol. Cell. Biochem. 2016, 414, 37–46.
- Das, S.; Ferlito, M.; Kent, O.A.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA Regulates the Mitochondrial Genome in the Heart. Circ. Res. 2012, 110, 1596–1603.
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c Regulates the Mitochondrial Genome, Bioenergetics, and Propensity for Heart Failure In Vivo. PLoS ONE 2014, 9, e96820.
- Mancini, M.; Saintigny, G.; Mahé, C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. MicroRNA-152 and -181a participate in human dermal fibroblasts senescence acting on cell adhesion and remodeling of the extra-cellular matrix. Aging 2012, 4, 843–853.
- Aschrafi, A.; Schwechter, A.D.; Mameza, M.G.; Natera-Naranjo, O.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 Regulates Local Cytochrome c Oxidase IV mRNA Levels and Oxidative Phosphorylation in the Axons of Sympathetic Neurons. J. Neurosci. 2008, 28, 12581–12590.
- Aschrafi, A.; Kar, A.N.; Natera-Naranjo, O.; MacGibeny, M.A.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery. Cell. Mol. Life Sci. 2012, 69, 4017–4027.
- Jagannathan, R.; Thapa, D.; Nichols, C.E.; Shepherd, D.L.; Stricker, J.C.; Croston, T.L.; Baseler, W.A.; Lewis, S.E.; Martinez, I.; Hollander, J.M. Translational Regulation of the Mitochondrial Genome Following Redistribution of Mitochondrial MicroRNA in the Diabetic Heart. Circ. Cardiovasc. Genet. 2015, 8, 785–802.
- Knezevic, I.; Patel, A.; Sundaresan, N.R.; Gupta, M.P.; Solaro, R.J.; Nagalingam, R.S.; Gupta, M. A Novel Cardiomyocyte-enriched MicroRNA, miR-378, Targets Insulin-like Growth Factor 1 Receptor. J. Biol. Chem. 2012, 287, 12913–12926.
- Giuliani, A.; Prattichizzo, F.; Micolucci, L.; Ceriello, A.; Procopio, A.D.; Rippo, M.R. Mitochondrial (Dys) Function in Inflammaging: Do MitomiRs Influence the Energetic, Oxidative, and Inflammatory Status of Senescent Cells? Mediat. Inflamm. 2017, 2017, 1–11.
- Ji, G.; Lv, K.; Chen, H.; Wang, T.; Wang, Y.; Zhao, D.; Qu, L.; Li, Y. MiR-146a Regulates SOD2 Expression in H2O2 Stimulated PC12 Cells. PLoS ONE 2013, 8, e69351.
- Rippo, M.R.; Olivieri, F.; Monsurrò, V.; Prattichizzo, F.; Albertini, M.C.; Procopio, A.D. MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Exp. Gerontol. 2014, 56, 154–163.
- Yan, K.; An, T.; Zhai, M.; Huang, Y.; Wang, Q.; Wang, Y.; Zhang, R.; Wang, T.; Liu, J.; Zhang, Y.; et al. Mitochondrial miR-762 regulates apoptosis and myocardial infarction by impairing ND2. Cell Death Dis. 2019, 10.
- Hackl, M.; Brunner, S.; Fortschegger, K.; Schreiner, C.; Micutkova, L.; Mück, C.; Laschober, G.T.; Lepperdinger, G.; Sampson, N.; Berger, P.; et al. miR-17, miR-19b, miR-20a, and miR-106a are down-regulated in human aging. Aging Cell 2010, 9, 291–296.
- Giuliani, A.; Cirilli, I.; Prattichizzo, F.; Mensà, E.; Fulgenzi, G.; Sabbatinelli, J.; Graciotti, L.; Olivieri, F.; Procopio, A.D.; Tiano, L.; et al. The mitomiR/Bcl-2 axis affects mitochondrial function and autophagic vacuole formation in senescent endothelial cells. Aging 2018, 10, 2855–2873.
- Diniz, G.P.; Lino, C.A.; Guedes, E.C.; do Nascimento Moreira, L.; Barreto-Chaves, M.L.M. Cardiac microRNA-133 is down-regulated in thyroid hormone-mediated cardiac hypertrophy partially via Type 1 Angiotensin II receptor. Basic Res. Cardiol. 2015, 110, 49.
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.-L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007, 13, 613–618.
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078.
- Wang, K.; Lin, Z.-Q.; Long, B.; Li, J.-H.; Zhou, J.; Li, P.-F. Cardiac hypertrophy is positively regulated by MicroRNA miR-23a. J. Biol. Chem. 2012, 287, 589–599.
- Li, M.; Wang, N.; Zhang, J.; He, H.-P.; Gong, H.-Q.; Zhang, R.; Song, T.-F.; Zhang, L.-N.; Guo, Z.-X.; Cao, D.-S.; et al. MicroRNA-29a-3p attenuates ET-1-induced hypertrophic responses in H9c2 cardiomyocytes. Gene 2016, 585, 44–50.
- Pan, Z.; Sun, X.; Shan, H.; Wang, N.; Wang, J.; Ren, J.; Feng, S.; Xie, L.; Lu, C.; Yuan, Y.; et al. MicroRNA-101 Inhibited Postinfarct Cardiac Fibrosis and Improved Left Ventricular Compliance via the FBJ Osteosarcoma Oncogene/Transforming Growth Factor-β1 Pathway. Circulation 2012, 126, 840–850.
- Zhao, X.; Wang, K.; Liao, Y.; Zeng, Q.; Li, Y.; Hu, F.; Liu, Y.; Meng, K.; Qian, C.; Zhang, Q.; et al. MicroRNA-101a inhibits cardiac fibrosis induced by hypoxia via targeting TGFβRI on cardiac fibroblasts. Cell. Physiol. Biochem. 2015, 35, 213–226.
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. miR-29 Is a Major Regulator of Genes Associated with Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294.
- Maurer, B.; Stanczyk, J.; Jüngel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.W.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62, 1733–1743.
- Du, B.; Ma, L.-M.; Huang, M.-B.; Zhou, H.; Huang, H.-L.; Shao, P.; Chen, Y.-Q.; Qu, L.-H. High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 2010, 584, 811–816.
- Liu, Y.; Taylor, N.E.; Lu, L.; Usa, K.; Cowley, A.W.; Ferreri, N.R.; Yeo, N.C.; Liang, M. Renal Medullary MicroRNAs in Dahl Salt-Sensitive Rats. Hypertension 2010, 55, 974–982.
- Wu, Q.; Xu, T.; Liu, Y.; Li, Y.; Yuan, J.; Yao, W.; Xu, Q.; Yan, W.; Ni, C. MiR-1224-5p mediates mitochondrial damage to affect silica-induced pulmonary fibrosis by targeting BECN1. Int. J. Mol. Sci. 2017, 18, 2357.
- Zhong, X.; Chung, A.C.K.; Chen, H.-Y.; Meng, X.-M.; Lan, H.Y. Smad3-Mediated Upregulation of miR-21 Promotes Renal Fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681.
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984.
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030.
- Srinivasan, H.; Das, S. Mitochondrial miRNA (MitomiR): A new player in cardiovascular health. Can. J. Physiol. Pharmacol. 2015, 93, 855–861.
- Banavath, H.N.; Roman, B.; Mackowski, N.; Biswas, D.; Afzal, J.; Nomura, Y.; Solhjoo, S.; O’Rourke, B.; Kohr, M.; Murphy, E.; et al. miR-181c Activates Mitochondrial Calcium Uptake by Regulating MICU1 in the Heart. J. Am. Heart Assoc. 2019, 8, e012919.
- Stocum, D.L. Regenerative Biology and Medicine; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2012; ISBN 9780123848604.
- Carrer, M.; Liu, N.; Grueter, C.E.; Williams, A.H.; Frisard, M.I.; Hulver, M.W.; Bassel-Duby, R.; Olson, E.N. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378. Proc. Natl. Acad. Sci. USA 2012, 109, 15330–15335.
- Sousounis, K.; Baddour, J.A.; Tsonis, P.A. Aging and Regeneration in Vertebrates. Curr. Top. Dev. Biol. 2014, 108, 217–246.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z. Bin Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.