Cyclic adenosine monophosphate (cAMP) is a small molecule that acts as a second messenger in mediating intracellular signal transduction.
- connexin,cAMP
1. Introduction
Cyclic adenosine monophosphate (cAMP) is a small molecule that acts as a second messenger in mediating intracellular signal transduction [1,2]. Early studies indicated that cAMP signaling depends primarily on its activation of protein kinase A (PKA) [3]. However, four other key mammalian effector protein families have also been shown to be direct targets of cAMP, including the exchange protein activated by cAMP (EPACs1 and 2), the cyclic nucleotide gate channels, proteins containing Popeye domains, and a cyclic nucleotide receptor involved in sperm function [4]. Notably, cancer research has primarily focused on PKA and EPAC [5].
The cAMP/PKA signaling pathway is involved in the invasion, migration, adhesion, clonal growth, and other malignant phenotypes of cancer cells, including glioblastoma, ovarian cancer, colorectal cancer, breast cancer, and also pituitary tumors [6,7,8,9,10]. In particular, PKA phosphorylation of vasodilator-stimulated phosphoprotein (VASP) enhances the invasion and metastasis of esophageal squamous cells [11]. In addition, a related study on CIP4 PKA phosphorylation showed its regulation of cancer metastasis [12]. Moreover, TGFβ/PKA was found to be reportedly involved in colon cancer metastasis [13]. Therefore, PKA appears to be pivotal for malignant transformation.
Research on cAMP signaling in the cancer field has primarily aimed at identifying effective therapeutic targets for cancer [2,14]. However, there are several important studies focusing on the cross-talk among cAMP, Cx, and gap junction intercellular communication (GJIC). Connexins (Cxs), with 21 subtypes in humans, form both gap junctions and hemichannels that mediate the communications between two adjacent cells and between intracellular, and extracellular microenvironments, respectively [15,16,17,18]. Cxs are regulated by the cAMP signaling pathway, whereas Cx channels mediate the transfer of cAMP between cells via GJIC and in some circumstances, release to the extracellular milieu via Cx hemichannels. The significance of this phenomenon in cancer biology has been recently highlighted by several independent studies. Research conducted previously primarily focused on the mechanisms underlying the tumor-suppressor function of Cxs within a single cell, but more recently, attention has shifted toward an understanding of role of Cx channels in cAMP signaling in relation to the entire cell population [19,20]. One recent study demonstrated that cAMP exchange between cells via gap junctions limits cell cycle progression [21], proposing a potentially unifying model that could explain how gap junctions modulate tumor progression. Hepatic epithelial cells are instead very sensitive to the levels of both cAMP and Cxs. For example, increased functionality of the gap junctions or elevated levels of cAMP have been shown to reduce the tumorigenicity of HCC cells [22]. However, these mechanisms, which are pivotal to tissue homeostasis, require further investigation.
Cxs are direct targets of cAMP/PKA signaling [23], with the assembly of Cx43 channel clusters between cells being modified by cAMP signaling [24]. The cAMP/PKA pathway regulates the expression and phosphorylation of Cxs. Persistent hyperglycemia reduces Cx36 mRNA levels in insulin-secreting cells in a rat model of continuous glucose infusion, and blocking the cAMP/PKA pathway attenuates this effect [25]. The cAMP/PKA pathway phosphorylates Cxs, and like in the zebrafish retina, Cx35/36 is directly phosphorylated by PKA at serine 110 and 276/293 to regulate photoreceptor coupling [26]. PKA directly phosphorylates Cx43 in rats, forming a Cx43 gap junction regulated by follicle-stimulating hormone [27], whereas the PKA–ezrin–Cx43 axis directly regulates GJIC in the human placenta to trigger trophoblast cell fusion [28]. Therefore, whereas Cx channels mediate the intercellular transfer of cAMP, they are also regulated by it.
2. Regulation of Cx Expression by cAMP/PKA Signaling in Cancer Cell Growth and Primary Cancer Progression
Cx expression has long been correlated with the occurrence, progression, and metastasis of cancer. Sixty years ago, Loewenstein and colleagues observed the loss of intercellular coupling in numerous cancers, and Cxs have been characterized as tumor suppressors in several cancer subtypes [19,29]. The overexpression of Cxs has been shown to inhibit primary tumor growth and progression in pancreatic cancer cells and breast cancer [19], as is the case for Cx43 in colorectal cancer cells [30]. However, recent studies have shown that Cx expression is tissue- and stage-specific in cancer. For example, the expression profile of Cx43 in breast cancer is largely dependent on the intrinsic subtype of breast cancer, but the majority of breast cancer cells exhibit reduced Cx43 expression or its aberrant localization in the cytoplasm [31]. In addition to those in the primary tumor, the changes in Cx expression are directly correlated with cancer metastasis. The metastasis of tumor cells from a primary site to distant foci is perhaps the most critical stage in cancer progression, as this is what typically leads to terminal outcomes for patients. In this phase, the role of Cxs is typically opposite, as the upregulation of Cx expression is most often positively associated with metastatic cancer, such as the role of Cx43/Cx26/Cx30 in metastatic breast cancer and prostate cancer bone metastasis [32,33,34,35,36]. Furthermore, Cxs are involved in entry (intravasation) and exit (extravasation) from the bloodstream [32,37,38], and several Cxs, including Cx43/Cx26/Cx30, have been implicated in the invasion of remote tissues, like prostate cancer bone metastasis (Cx43), breast cancer lymphatic vessel invasion (Cx26), and breast cancer and melanoma brain metastasis (Cx43 and Cx26) [32,33,34].
cAMP signaling plays a role in cancer cell migration; however, it does not alter Cx43 and Cx32 expression, but rather upregulates Cx26 [39]. In addition to Cx expression, it is noteworthy that cell movement depends on the formation of gap junction channels [40], especially the channels formed by Cx26 [41]. This might partially explain the response of Cx26 expression to cAMP signaling.
In several systems, cAMP via PKA appears to be critical in regulating Cx expression, although whether it exerts positive or negative effects appears to depend on the tissues and tissue-specific Cx expression profile. However, these studies were not conducted on cancer cells. For example, continuous hyperglycemia results in a decline in the mRNA levels of rat Cx36 (rCx36) in insulin-secreting cells in rats administered a continuous glucose infusion, an effect that is blocked by the inhibition of cAMP/PKA signaling [25]. In contrast, prostaglandins have been shown to induce the expression of rat Cx43 (rCx43), which can be reversed by the adenylate cyclase inhibitor SQ22536 or the PKA inhibitor H89 in mesangial and bladder smooth muscle cells [42].
The cAMP response element-binding protein (CREB) can directly regulate the transcription of Cx43 by forming complexes with nuclear factor kappa-B (NF-κB) and CREB-binding protein (CBP), the latter two being important effectors of cAMP [43]. The modulation of CREB enhances rCx43 expression [43]. Conversely, inducible cAMP early repressor I decreases rCx36 expression in pancreatic islets in rats [25]. Moreover, oxytocin regulates mCx43 expression by activating cAMP/PKA signaling and enhancing the expression of nuclear factor kappa-light-chain enhancer in mouse embryonic stem cells [44] (Figure 1). Although these studies have been done with non-cancer cells, a similar mechanism could be adopted by certain types of cancer cells. Moreover, the NF-kB/CBP/CREB complex can regulate the expression of target genes both positively and negatively [45]; thus, studies that address both of these circumstances should be considered. It would be interesting to explore the effect of the cAMP/NF-κB/Cx axis in cancer apoptosis.
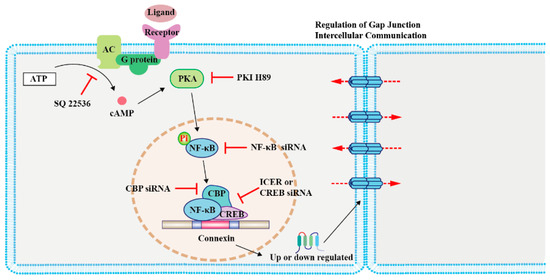
Figure 1. Regulation of connexin (Cx) expression by cAMP/PKA signaling in modulating cancer cell growth and primary cancer progression. The schematic illustrates the mechanism involved in the cAMP signaling-mediated regulation of Cx levels. Activation of the G-protein-coupled receptor on the cell membrane by ligands leads to cAMP synthesis by AC. cAMP diffuses into the cell and activates its effector protein PKA, which phosphorylates NF-κB. Subsequently, NF-κB, CBP, and CREB form a complex to regulate the transcription of Cxs [25,42,43,44]. Abbreviations: AC, adenylyl cyclase; PKA, protein kinase A; NF-κB, nuclear factor kappa-B; CBP, CREB-binding protein; CREB, cAMP response element-binding protein; PKI, PKA inhibitor; and ICER, inducible cAMP early repressor I.
3. cAMP/PKA Signaling Mediates the Phosphorylation of Cxs to Promote Cancer Cell Migration and Malignant Transformation
The functional regulation of GJIC, especially Cx phosphorylation, is necessary for cancer cell migration [46]. The phosphorylation of rCx43 at S255, S279, and S282 has been reported, which either opens or closes the gap junction [47,48,49]. Cx43 contains multiple phosphorylation sites regulated by kinase-mediated signaling pathways that interact with other proteins, including PKA [49,50]. A previous study indicated that stimulating the cAMP pathway regulates hemichannel switching, as well as endocrine and paracrine signaling [51]. Another study showed that dipyridamole induces cAMP production to post-translationally modify bovine Cx43 (bCx43), resulting in increased gap junction coupling of endothelial cells [52].
Interestingly, Cx43 phosphorylation by PKA seemingly does not directly affect the channel but has been linked to trafficking. rCx43 phosphorylation can be influenced by PKA localization and by other factors. For example, the tight junction protein 1 (ZO-1) binds both PKA and Cx43, bringing the kinase in proximity to its targets on Cx43, which leads to gap junction channel opening [53]. In fact, separation from ZO-1 is necessary for gap junction closure [53]. It was also determined that the complex formed by ezrin, rCx43, and ZO-1 brings PKA in proximity and phosphorylates rCx43, promoting the opening of the gap junction channels (Figure 2) [54]. In contrast, PKA inhibition diminishes rCx43 phosphorylation [55]. These data indicated that the cAMP/PKA pathway is instrumental in the phosphorylation of rCx43.
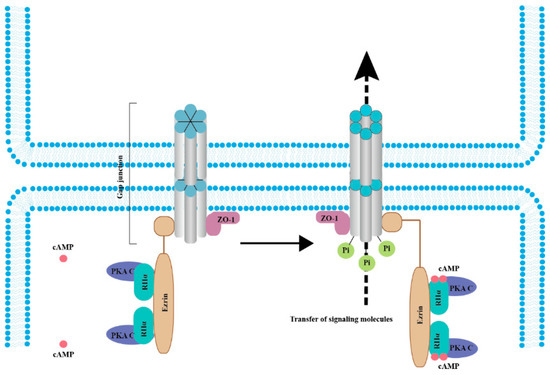
Figure 2. cAMP/PKA signaling mediates the phosphorylation of connexins (Cxs) to promote cancer cell migration and malignant transformation. In the resting state (left panel), the whole complex consists of Cx, ZO-1, Ezrin, and a PKA pool anchored by Cx-bound ezrin. When cAMP levels increase (right panel), PKA regulatory subunits bind to four cAMP molecules, resulting in activated PKA catalytic subunits, increased Cx phosphorylation, and gap junction-mediated intercellular coupling [26,53,54,55]. Abbreviations: RIIα, PKA regulatory subunits and PKA C, PKA catalytic subunits.
The accelerated proliferation of cancer cells is reportedly associated with Cx43 phosphorylation. Phosphorylated Cx43 is upregulated during hyperplasia, atypical hyperplasia, fibroadenoma, and especially in invasive cancer [56]. Similarly, human gliomas exhibit the significant overexpression of phosphorylated Cx43 [57]. Together, these data indicate that the malignant proliferation of cancer cells is related to the phosphorylation of Cx43. However, in many of these studies, the particular kinases mediating the phosphorylation reaction have not been identified. Notably, PKC, extracellular regulated protein kinases, and tyrosine kinases like c-src can phosphorylate Cx43, which generally causes the closure of the channels [58]. In non-cancer, specifically in the nervous system, the phosphorylation of goldfish Cx35 (gCx35) is associated with the dynamic control of gap junction coupling in the zebrafish Mauthner neuron [59]. Furthermore, this has been observed in the zebrafish retina, where cAMP activates the channels and mediates the activity-dependent potentiation of coupling; PKA activity plays a key role in this process [26]. Therefore, cAMP-activated PKA phosphorylates Cx to regulate GJIC. Cx35/36 phosphorylation by PKA seems to directly affect the probability of channel opening. However, caution is needed in interpreting these results, as the effects observed by blocking PKA do not necessarily imply direct phosphorylation of the Cx protein, unless this is actually demonstrated.
4. Gap Junction Channels Mediate Transfer of cAMP
In addition to considering Cx as targets of cAMP-mediated signaling, it is also relevant to consider the roles of Cxs and GJIC in its propagation. However, it is crucial to consider that not all Cxs form channels with similar permeability for cAMP. For example, cAMP permeability is much higher through rCx43 than Cx50 channels expressed in HeLa cells [60]. Furthermore, rCx43, mCx46, and Cx50 present a different permeability for cAMP in lens cells [60]. In a more comprehensive study using a more indirect reporter of cAMP transfer, the order of cAMP permeabilities through different Cxs is as follows: Cx43 > Cx26 > Cx45 = Cx32 > Cx47 > Cx36 [61]. However, direct measurements of cAMP transfer by Cxs expressed in oocytes show that Cx26 > Cx43 ~ Cx32 (Nicholson, personal communication). For example, cAMP transfer between cells mediated by Cx43 modifies the expression of key effector molecules and genes in interconnected osteosarcoma cells [62].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13010058