A diverse range of linear polysiloxane-based ionic polymers that are hydrophobic and highly flexible can be obtained by substituting the polymers with varying amounts of ionic centers. The materials can be highly crystalline solids, amorphous soft solids, poly(ionic) liquids or viscous polymer liquids.
- polysiloxane
- ionomer
- polyelectrolyte
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
1.1. A Brief History of Polysiloxanes, Including an Overview of Their Properties
Since the seminal work of Frederic Kipping on organosilanes, commencing more than 100 years ago [1], polysiloxanes and silicones have been used for an enormous range of applications [2–11]; they pervade our lives. The applications include electrical insulation [2–4], water repellents for leather [5], adhesives [6,8,11], surfactant agents for both foaming and anti-foaming [7,9], and personal care products [10]. The continued popularity of these materials can be attributed to their high stability to heat, light and many classes of chemicals [12–14], as well as their flexibility, and ease of modification of other mechanical properties. Neat polydimethylsiloxanes (PDMSs) are structurally flexible due to the low torsional energy barrier around their Si-O-Si bonds (0–5 kJ/mol [15,16]) and the related glass transition temperatures (Tg) that are lower than those of linear polyacrylates of a similar molecular weight. The ability to synthesize polysiloxanes by polycondensation reactions [13] between cyclic oligosiloxanes and functionalized dichlorosilanes provides a plethora of materials with different chemical structures and mechanical behaviors that can be tuned for different applications. For example, the commonly employed Karstedt’s catalyst [17,18] can be used to convert cyclic oligosiloxanes from liquids at room temperature into flexible plastic and gel-like materials that have been used as platforms for optoelectronics [19,20]. With appropriate polar functional groups, polysiloxanes become somewhat hydrophilic, making them useful as adhesives and sealants [11] despite the parent polymers being highly hydrophobic. For example, aminopropyl dimethicone and amodimethicone [21], classes of polysiloxane copolymers with aminopropyl and dimino-containing chains, form emulsions in water. Also, the amino groups can be protonated using mild acids, and even form crosslinks with diacids; in both cases, ion pairs, that alter the local polarity of the polymers, are produced [22]. Although the use of amodimethicone and other polysiloxane-based copolymers in cosmetic [23] and textile products [24] appeared in the 1980s, functionalized ionic polysiloxanes have been introduced more recently. Recent developments in the field of polysiloxanes that exploit non-covalent (especially ionic) cross-links is the focus of this mini-review. Also, the increasing number and type of applications of polysiloxane-based ionic polymers, in fields as diverse as fuel cells and electrolytic media for batteries [25,26], make a review of these materials especially pertinent at this time.
1.2. Ionic Polymers and Polysiloxanes
Ionomers are ionic polymers with a small fraction (defined formally by the International union of pure and applied chemistry (IUPAC) as 10%) of monomers containing charged groups. Ionic polymers with a higher fraction of monomers containing charged groups are defined by IUPAC as polyelectrolytes.
As a result of the charged centers, ionomers have a propensity to form microphases consisting of small ionic clusters (approximately 1–5 nm in size [27]) embedded in the amorphous regions. The physical properties of the polymers depend on the ion content because the strength of the electrostatic interactions between ion pairs exceeds the dispersive polymer–polymer chain polysiloxane interactions. When ionomers are dissolved in low dielectric solvents, the ionic interactions remain a dominant factor, but they are less important in solvents of high polarity. The distribution of the sizes is due to numerous factors related to the polymer architecture [27]: molecular weight, type of pendant groups, polymer backbone type, random versus block substituent placement, etc. One disadvantage of ionic aggregation is the lowering of conductivity relative to polymers without ionic clustering [28].
IUPAC provides only a qualitative definition of polyelectrolytes, and modifications of them include ionic polymers (ionomers) and their interactions with solvents [29,30]. Because this review focuses on neat ionic polymer melts, the original limit suggested by IUPAC of 10% by mole for the ion content will be used here to distinguish ionomers from polyelectrolytes. The greater number of non-covalent ionic interactions among the constituents of polyelectrolytes also helps to distinguish them from ionomers. Many ionomers are semi-crystalline solids whose packing can be discerned from analyses of X-ray diffraction data. In addition, when monomers of ionic liquids are polymerized, a special category of polyelectrolytes, called poly(ionic) liquids (or polymeric ionic liquids), is formed; many poly(ionic) liquids remain liquids at temperatures below 100 °C [31–33]. These materials have been studied for their potential to combine the benefits of polyelectrolytes (i.e., flexible macromolecular architectures) and ionic liquids (i.e., high thermal stabilities and ionic conductivities) [34,35].
2. Types of Polysiloxane-Based Ionic Polymers
2.1. Polyelectrolytes Where All Monomers Except the Terminal Group Contain an Ion
Polysiloxane-based polyelectrolytes with inorganic counterions. The flexibility of the polysiloxane polymer chains can be decreased drastically by increasing the frequency of ionic cross-linker sites [31,36–40]. Polyelectrolyte examples of this effect, shown in Figure 1A, have been studied by Kaneko and coworkers [36–39,41]. This general structure is related to the polyelectrolyte ionomer (with far fewer ionic groups), aminopropyl dimethicone. Another key structural difference between the structures in Figure 1A and aminopropyl dimethicone is the presence of a hydroxyl group in the former instead of a methyl group in the siloxane monomer. Each of the aminopropyl-trimethoxysiloxane monomeric units (APTMOS) used to form the resulting siloxane-based polyelectrolytes (PAPS) in Figure 1 contains a charged ammonium ion with either chloride or nitrate (PAPS-Cl or PAPS-NO3, respectively) as the counterion. Neither the average molecular weight nor the polydispersity was reported for either the PAPS-Cl or PAPS-NO3; bulk characterization consisted of x-ray diffraction (XRD), transmission and scanning electron microscopies, and nitrogen porosimetry. Due to the high density of ion pairs and hydroxyl groups along the polymer chains, the formation of highly ordered structures is not surprising. XRD data showed that the chains of both PAPS-Cl and PAPS-NO3 pack in hexagonally ordered arrays that implicate somewhat extended polymer chains.
The highly hydrophilic and hygroscopic nature of the PAPS-Cl or PAPS-NO3 salts also follows from the prevalence of the multiple ion pairs. The effects on the packing of the chloride salt, PAPS-Cl, was explored by dissolving it in water first and then recording the x-ray diffractograms at various stages of drying. Thus, it was found that the slow removal of water by drying in air does not change the overall packing motif.
PAPS-Cl and PAPS-NO3 are not soluble in low polarity, aprotic solvents such as chloroform and dichloromethane. Curiously, the polyelectrolytes can be dissolved in DMSO but not in some other polar (and some protic) solvents such as ethanol, methanol, acetone and N,N-dimethylformamide.
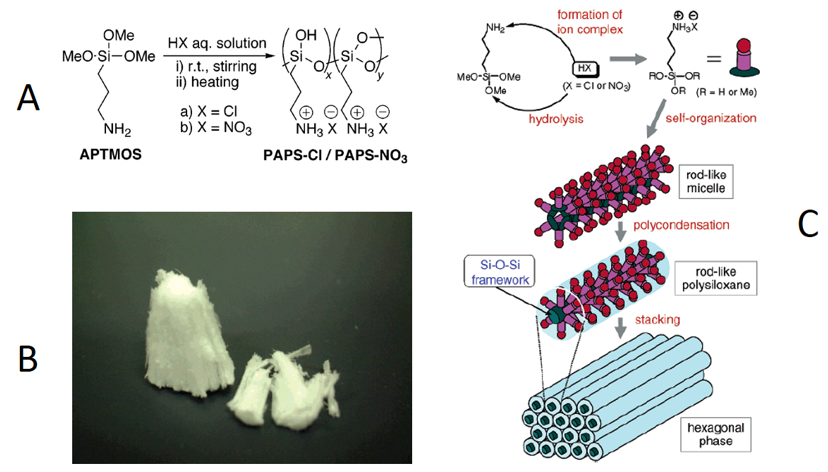
Figure 1. Neat polysiloxane-based polyelectrolytes with propylammonium chloride or nitrate pendant groups [36]. (A) The synthetic route to PAPS-Cl and PAPS-NO3. (B) Photograph of neat PAPS-Cl. (C) Cartoon representation of the ordered layers based on x-ray diffraction (XRD) data. Reprinted with permission from [36]. Copyright (2004) (American Chemical Society).
Polysiloxane-based polyelectrolytes with organic counterions. Organic counterions, such as long-chained carboxylates, in polysiloxane-based polyelectrolytes introduce another structural variable: an increased importance of London dispersion forces. Thus, Kaneko and coworkers [36,37] have combined PAPS with octanoate as the counterion (PAPS-C8). The neat material is a hygroscopic powder, similar in appearance and packing to PAPS-Cl and PAPS-NO3. XRD indicates hexagonal packing, again, but with a wider cylindrical diameter due to the alkyl chains. Kaneko and coworkers [36] mention that PAPS with carboxylate counterion chains longer than octanoate (N.B., decanoate, dodecanoate, and tetradecanoate) do not maintain hexagonal packing, probably due to increased London dispersion interactions and entropic disorder. Also, PAPS with carboxylate chains shorter than octanoate (N.B., hexanoate and butanoate) remained liquids at room temperature; they are polymeric ionic liquids. Additionally, carboxylates as counterions make the PAPS electrolytes more hydrophobic; whereas PAPS-C8 is soluble in ethanol, neither PAPS-Cl nor PAPS-NO3 can be dissolved in this alcohol.
The correlation between the bulk properties of polyelectrolytes and the length of the alkyl chain in the counterion is interesting, and has been found in other examples involving polyelectrolyte-surfactant complexes [42–45]. Polyacrylate-based polycations with hexanoate and shorter alkyl carboxylate anions are also ionic liquids at room temperature, but form ordered structures when the carboxylates are decanoate or longer.
Polysiloxane-based polyelectrolytes with a polymeric counterions. To the best of our knowledge, there is only one report of a polysiloxane-based polyelectrolyte in which all monomeric units contain an ion pair [37]: PAPS-Cl has been combined with another polymeric polyelectrolyte, sodium polyacrylate (molecular weight of 250 KDa). The resultant nano-composite was heterogeneous because the two polymers exhibited minimal interactions. Their nearly complete separation allowed the rod-like polysiloxane to retain its hexagonal packing while being surrounded by layers of sodium polyacrylate.
Polysiloxane-based poly(ionic) liquids/polymer ionic liquids. Due to their flexibility, polysiloxane-copolymer chains have been shown to form polymeric ionic liquids (PILs). Both of the PILs shown in Scheme 1 [31,32] have pendant groups that lack sufficient non-covalent interactions to be in a semi-crystalline state at room temperature. In the PILs designed by Jourdain and coworkers [31], a triazolium bis(trifluoromethyl)sulfonamide ion pair is generated through click chemistry using a copper(I)-catalyzed azide-alkyne cycloaddition. The presence of the oligomeric ethylene oxide group promotes packing of the chains, although the glass transition temperature is still only ca. −62 °C; by comparison, the glass transition temperature of neat, high molecular weight PDMS is −123 °C [12]. A similar result was found for the PILs made by Bocharova and coworkers [46], where the length of the alkyl chain is too short to generate lamellar packing that is stable at room temperature. The trends observed for both PILs can be summarized by a correlation of the monomer molar volume and glass transition temperature. For PILs with a flexible backbone, a smaller molar volume of the monomer (including the counterion) results in a higher glass transition temperature. This observation may explain why the polyelectrolyte salts of Kaneko and coworkers [36,38] did not form polymeric ionic liquids—their molar volumes are smaller. Lastly, one assumption implicit in this correlation is that the counterions of the PILs do not contribute significantly to the non-covalent interactions (i.e., bis(trifluoromethane)sulfonimide (TFSI) and the halide anions do not aggregate or phase separate into structures similar to those found for organic anions such as long-chain carboxylates). More examples will be needed to test this hypothesis adequately.
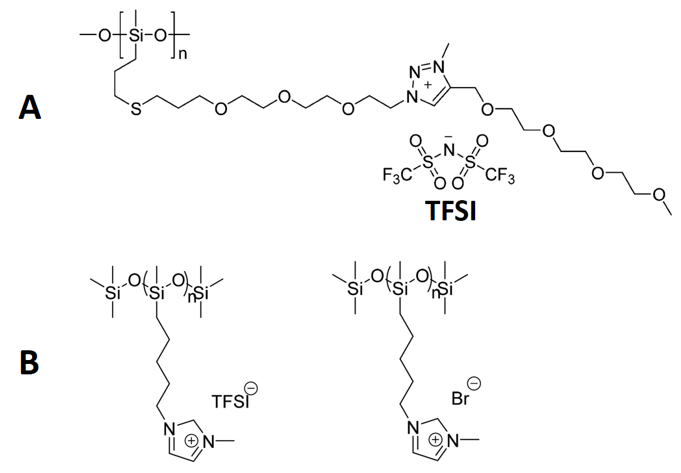
Scheme 1. Two examples of potentially important polymeric ionic liquids [31,32]: (A) a TFSI-based polymeric ionic liquid (PIL) and (B) an imidazolium-based PIL. Reprinted with permission from [31,32]. Copyright (2016 and 2017) (American Chemical Society).
2.2. Polysiloxane-Based Polyelectrolytes with Some Monomers not Containing Ion Pairs
Influence of lowering the ion pair content. A decrease in the amount of non-covalent crosslinkers does not always translate to a decrease in their influence on the physical state of an ionic polymer; a broad range of possible materials can still be formed [40,47,48]. Much of the intermolecular interactions that influence the physical state of the material is now delegated to the pendant groups and counterion. Thus, a polysiloxane-based imidazolium bromide polyelectrolyte salt remains a liquid at room temperature even with 25% by mole of imidazolium bromide ion content. Beyond noting that this material is a Newtonian liquid at room temperature, not much else is known about the physical texture of the polyelectrolyte. Also using an imidazolium bromide (NL) ion pair, Zuo and coworkers [40] in 2017 synthesized polyelectrolytes (PNL1-n) with differing NL grafting densities using thiol-ene chemistry (Figure 2A). Ranging from 16% for PNL1-1 to 85% for PNL1-5 (Figure 2), the grafting density was calculated using the weight average molar mass provided. Surprisingly, at a higher grafting density, the authors found that the resulting polyelectrolyte remains a viscous liquid. The authors attribute this result to the disruption of entanglements that occurs for PNL1-4 and PNL1-5, but not for PNL1-3. This is interesting, as the polysiloxane copolymer with mercaptopropyl pendant groups (PMMS) is a liquid as noted by the rheological data. Thus, there appears to be an optimal grafting density range that results in a phase transition. An unanswered question in this work involves the 365 nm wavelength used to photo-initiate the thiol-ene click chemistry This radiation can also induce the formation of disulfide linkages in primary alkyl thiols [49], and they may also contribute to the different physical states of the materials.
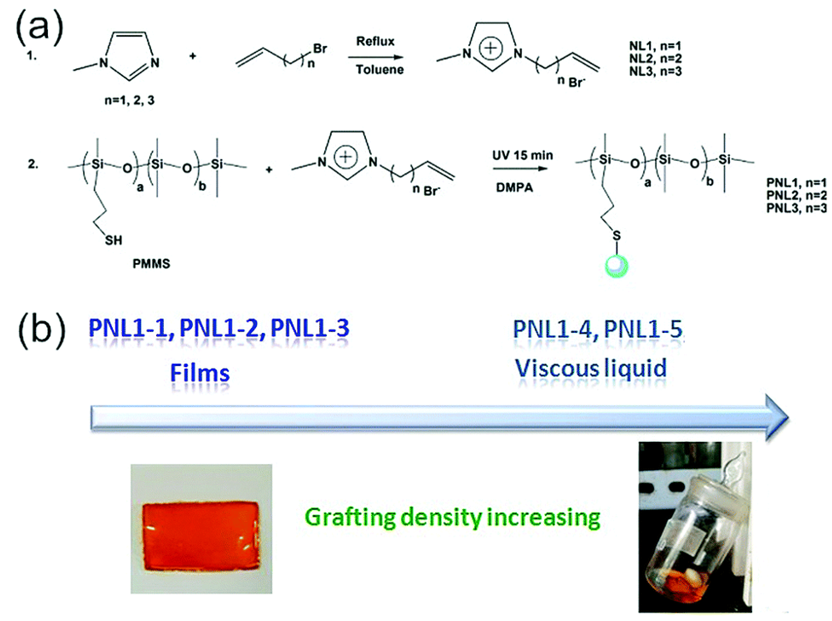
Figure 2. (a) Reaction schemes and (b) pictures involving an imidazolium bromide polyelectrolyte system [40]. Reproduced from [40] with permission from the Centre National de la Recherche Scientifique (CNRS) and The Royal Society of Chemistry.
Influence of alkyl chain lengths at lowered ion content [50]. Many researchers are exploiting the influence of pendant groups on the bulk properties of ionic polymeric systems. The highly flexible nature of the linear polysiloxane homopolymers and the type and amount of a pendant group can be used to tailor physical properties, such as the glass transition temperature. Exploring different types of pendant groups can be used to introduce new properties not normally seen with linear polysiloxane homopolymers. A good example is the liquid-crystalline polyelectrolyte made by Wang and coworkers [50]. A set of polysiloxane copolymers, [PnSO3], were synthesized with two unique pendant groups: a phenylpropoxy sulfonate anion and a bulky cholesteric group. Amounts of these groups were varied and given as mole fractions of the sulfonate anion (Figure 3D). The lowest amount of sulfonate in the polysiloxane copolymer is denoted by n = 1 or [P1SO3] and the highest amount by n = 3 or [P3SO3]. The former was used to crosslink two different dications: hexane-1,6-diyl diisonicotinate (HPY) and (1,1′-bipenyl)-4,4′-diisonicotinate (BPy) (Figure 3C), making the networks far more rigid than those observed for the inorganic salt crosslinked systems mentioned in the previous section. The bulky cholesteric group provides the van der Waals ordering responsible for the liquid crystalline properties observed in the optical microscope images (Figure 3A,B). Due to greater intermolecular interactions from the pendant groups, the glass transition of the polyelectrolyte increases to 50 °C, with minimal influences from the crosslinkers [HPy] or [BPy]. It is also not surprising that that a smectic A or cholesteric mesophase is observed. They are typical of cholesteric liquid crystals [51]. One conclusion of note is that the thermal stability of the smectic A mesophase can be correlated with the amount of cholesteric pendant groups. The flexibility of the crosslinkers, HPy and BPy, also influences the thermal stability of the polyelectrolyte. Thermogravimetric analyses showed that the onset of pyrolysis of the organic component was 50 °C lower for [HPy][P1SO3] than for [BPy][P1SO3].
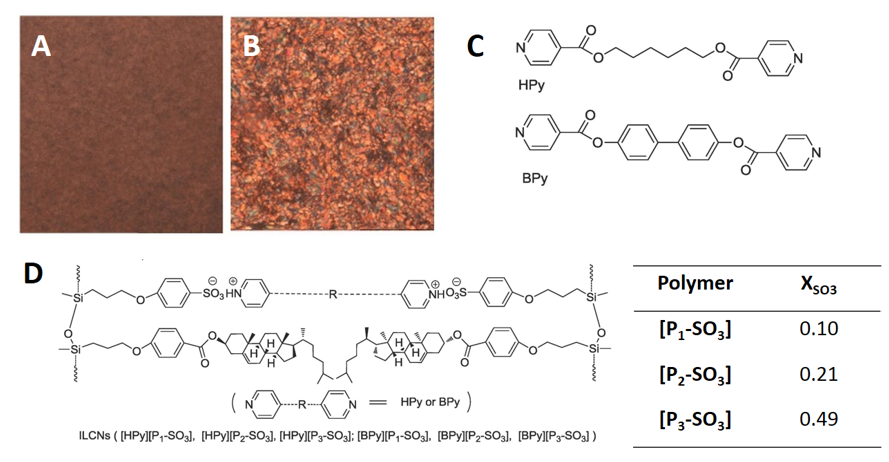
Figure 3. Optical microscope images of the liquid-crystalline polyelectrolyte salt [BPy][P1SO3] in its cubic phase at 80 °C (A) and smectic phase at 120 °C. (B). The structures of hexane-1,6-diyl diisonicotinate (HPY) and (1,1′-bipenyl)-4,4′-diisonicotinate (BPy) (C) and the final polyelectrolyte salts (D) are also shown [50]. Note: XSO3 is the molar fraction of each of the polymers. Adapted with permission from [50]. Copyright Elsevier (2018).
Polysiloxane-based ionomers. A key to categorizing the properties of ionic polymers is a determination of their propensity to form ion clusters (i.e., analysis of the regions rich and poor in ion pairs). Of the different types of ionic polymers, ionomers contain the fewest ion pairs and, for that reason, their bulk properties are the closest to those of unsubstituted polysiloxane homopolymers.
Microstructural properties of ionomers [48,52–56]. Because of their applications in single-ion conductivity, especially in materials containing lithium, understanding their microstructural properties has been a focus of ionomers research. Examples with polysiloxanes included here involve investigations of the microstructural consequences of changing the ion content of monomers with lithium perfluoroether sulfonate [48] and the nature of pendant groups [52–56]. One such example from the Colby group describes ionomers with halide counterions [52]. These ionomers cannot form covalent crosslinks. As a result, these ionomers remain liquids at room temperature [52]. Because polysiloxanes exhibit low glass transition temperatures, high hydrophobicity, and good thermal (Figure 4A) and chemical stability, Chen and coworkers targeted the ionomers for potential use as a media for ion transport [52]. The X-ray scattering data in Figure 4C show three unique peaks for this ionomer that can be attributed to the structural features in Figure 4B: the polysiloxane backbone (peak II), the oligo-ethylene oxide connecting groups that act as a plasticizer (peaks I and II), and the small quantity of phosphonium halide pendant groups that cluster (peak III). An interesting FT-IR method for following the progress of polysiloxane syntheses from non-ionic monomers has been reported recently [57,58]. It should be adaptable easily to follow the syntheses of ionic polymers reported here.
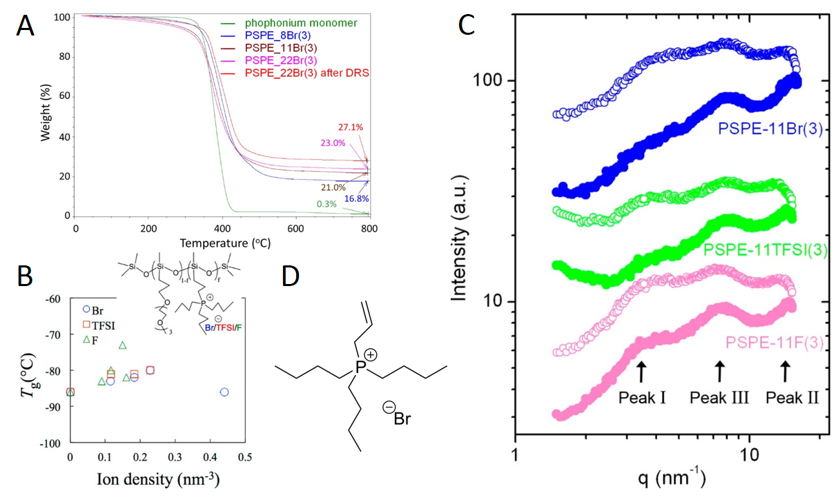
Figure 4. Polysiloxane-based ionomers with phosphonium halide pendant groups [52,53]. (A) Thermogravimetric (TGA) results for neat polysiloxane-based ionomers with phosphonium halide pendant groups. (B) Correlation between the glass transition temperature and ion density of the ionomers with various halide ions. (C) X-ray scattering spectra of polysiloxane-based ionomers with phosphonium bromide, bis(trifluoromethane)sulfonimide (TFSI), and fluoride pendant groups at varying temperatures; the filled and open circles are for data obtained at 25 and 125 °C, respectively. (D) Structure of the allyltributylphosphonium bromide monomer used for the TGA data in A. Reprinted with permission from [52,53]. Copyright (2013, 2014) (American Chemical Society).
References
- Thomas, N.R. Frederic Kipping—Pioneer in Silicon Chemistry: His Life & Legacy. Silicon 2010, 2, 187–193.
- Rochow, E.G. Methyl Aryl Silicones and Insulated Conductors and Other Products Utilizing the Same. U.S. Patent 2,258,222, October 7, 1941.
- Rochow, E.G. Methyl Silicones and Related Products. U.S. Patent 2,258,218, October 7, 1941.
- Hyde, J.F. Organo-Silicon Polymers and Method of Making Them. U.S. Patent 2,371,050, March 6, 1945.
- Currie, C.C. Leather Water Repellent. U.S. Patent 2,672,455, March 16, 1954.
- Currie, C.C.; Keil, J.W. Organopolysiloxane Adhesive and Pressure-Sensitive Adhesive Tape Containing Same. U.S. Patent 2,814,601, November 26, 1957; doi:10.1145/178951.178972.
- Rauner, L.A. Method for Reducing or Preventing Foam in Liquid Mediums. U.S. Patent 3,455,839, July 15, 1969.
- Hartlein, R. Epoxy Silane Coupling Agent. U.S. Patent 3,702,783, November 14, 1972; doi:10.1145/178951.178972.
- Cobb, V.S.; Rauscher, W.W.; Stanga, M.A.; Stevens, R.E.; Whitmarsh, R.H.; Wiese, K.D. Silicone Polyether Surfactants. U.S. Patent 5,830,970, November 3, 1998.
- Ahn, D.; Schulz, W.; Thompson, J. Silicone Compositions Comprising a Swollen Silicone Gel. U.S. Patent 9,243,113B2, January 26, 2016.
- Magalhäes, S.; Alves, L.; Medronho, B.; Fonseca, A.C.; Romano, A.; Coelho, J.F.J.; Norgren, M. Brief Overview on Bio-based Adhesives and Sealants. Polymers 2019, 11, 1685–1705, doi:10.3390/polym11101685.
- Kuo, C. M. Alex. Poly(Dimethylsiloxane). In Polymer Data Handbook; Mark, J., Ed.; Oxford University Press: New York, United States, 1999; pp. 411–435.
- Noll, W. CHAPTER 5—Preparation of Polyorganosiloxanes. In Chemistry and Technology of Silicones; Academic Press INC.: New York, NY, USA, 1968; pp. 190–245, doi:10.1016/B978-0-12-520750-8.50028-2.
- Noll, W. The Polymeric Organosiloxanes. In Chemistry and Technology of Silicones; Academic Press INC.: New York, NY, USA, 1968; pp. 246–331, doi:10.1016/B978-0-12-520750-8.50029-4.
- Smith, J.S.; Borodin, O.; Smith, G.D. A Quantum Chemistry Based Force Field for Poly(Dimethylsiloxane). Phys. Chem. B 2004, 108, 20340–20350, doi:10.1021/jp047434r.
- Weinhold, F.; West, R. The Nature of the Silicon-Oxygen Bond. Organometallics 2011, 30, 5815–5824, doi:10.1021/om200675d.
- Karstedt, B.D. Platinum Complexes of Unsaturated Siloxanes and Platinum Containing Organopolysiloxanes. U.S. Patent 3,775,452, November 27, 1973.
- Stochmal, E.; Strzezik, J.; Krowiak, A. Physicochemical and Catalytic Properties of Polysiloxane Network–Pt Systems. RSC Adv. 2017, 7, 26342–26360, doi:10.1039/C7RA00641A.
- Shanmuga sundar, D.; Sivanantha Raja, A.; Sanjeeviraja, C.; Jeyakumar, D. Highly Transparent Flexible Polydimethylsiloxane Films—A Promising Candidate for Optoelectronic Devices. Int. 2016, 65, 535–543, doi:10.1002/pi.5088.
- Neplokh, V.; Kochetkov, F.M.; Deriabin, K.V.; Fedorov, V.V.; Bolshakov, A.D.; Eliseev, I.E.; Mikhailovskii, V.Y.; Ilatovskii, D.A.; Krasnikov, D.V.; Tchernycheva, M.; et al. Modified Silicone Rubber for Fabrication and Contacting of Flexible Suspended Membranes of n-/p-GaP Nanowires with a Single-Walled Carbon Nanotube Transparent Contact. Available online: https://arxiv.org/abs/1910.13182 (accessed on 19 July 2020).
- CosIng—European Commission Database for Information on Cosmetics Substances and Ingredients. Available online: https://ec.europa.eu/growth/tools-databases/cosing/ (accessed on 19 July 2020).
- He, Y.; Zhao, H.; Yao, M.; Weiss, R.G. Complex New Materials from Simple Chemistry: Combining an Amino-Substituted Polysiloxane and Carboxylic Acids. Polym. Sci. Part A Polym. Chem. 2017, 55, 3851–3861, doi:10.1002/pola.28769.
- Disapio, A.; Fridd, P. Silicones: Use of Substantive Properties on Skin and Hair. J. Cosmet. Sci. 1988, 10, 75–89, doi:10.1111/j.1467-2494.1988.tb00004.x.
- Skinner, M.W.; Caibao, Q.; Grigoras, S.; Halloran, D.J.; Zimmerman, B.L. Fundamental Aspects of Aminoalkyl Siloxane Softeners by Molecular Modeling and Experimental Methods. Res. J. 1999, 69, 935–943, doi:10.1177/004051759906901209.
- Allen, M.H.; Wang, S.; Hemp, S.T.; Chen, Y.; Madsen, L.A.; Winey, K.I.; Long, T.E. Hydroxyalkyl-Containing Imidazolium Homopolymers: Correlation of Structure with Conductivity. Macromolecules 2013, 46, 3037–3045, doi:10.1021/ma302537f.
- Sanchez, J.Y.; Iojolu, C.; Alloin, F.; Guindet, J.; Lepretre, J.C. Fuel Cells—Proton Exchange Membrane Fuel Cells. Membranes: Non-Fluorinated. In Encyclopedia of Electrochemical Power Sources [Online]; Elsevier B.V. Posted December 8, 2009. https://www.sciencedirect.com/science/article/pii/B978044452745500887X (Accessed December 12, 2020)
- Zhang, L.; Brostowitz, N.R.; Cavicchi, K.A.; Weiss, R.A. Perspective: Ionomer Research and Applications. React. Eng. 2014, 8, 81–99, doi:10.1002/mren.201300181.
- Choi, U.H.; Lee, M.; Wang, S.; Liu, W.; Winey, K.I.; Gibson, H.W.; Colby, R.H. Ionic Conduction and Dielectric Response of Poly(Imidazolium Acrylate) Ionomers. Macromolecules 2012, 45, 3974–3985, doi:10.1021/ma202784e.
- Eisenberg, A.; Rinaudo, M. Polyelectrolytes and Ionomers. Bull. 1990, 24, 671, doi:10.1007/BF00300165.
- Zhang, Z.; Chen, Q.; Colby, R.H. Dynamics of Associative Polymers. Soft Matter 2018, 14, 2961–2977, doi:10.1039/c8sm00044a.
- Jourdain, A.; Serghei, A.; Drockenmuller, E. Enhanced Ionic Conductivity of a 1,2,3-Triazolium-Based Poly(Siloxane Ionic Liquid) Homopolymer. ACS Macro Lett. 2016, 5, 1283–1286, doi:10.1021/acsmacrolett.6b00761.
- Bocharova, V.; Wojnarowska, Z.; Cao, P.F.; Fu, Y.; Kumar, R.; Li, B.; Novikov, V.N.; Zhao, S.; Kisliuk, A.; Saito, T.; et al. Influence of Chain Rigidity and Dielectric Constant on the Glass Transition Temperature in Polymerized Ionic Liquids. Phys. Chem. B 2017, 121, 11511–11519, doi:10.1021/acs.jpcb.7b09423.
- Yuan, J.; Mecerreyes, D.; Antonietti, M. Poly(Ionic Liquid)s: An Update. Polym. Sci. 2013, 38, 1009–1036, doi:10.1016/j.progpolymsci.2013.04.002.
- Wang, Y.; Fan, F.; Agapov, A.L.; Yu, X.; Hong, K.; Mays, J.; Sokolov, A.P. Design of Superionic Polymers—New Insights from Walden Plot Analysis. Solid State Ionics 2014, 262, 782–784, doi:10.1016/j.ssi.2013.09.026.
- Wang, Y.; Sokolov, A.P. Design of Superionic Polymer Electrolytes. Opin. Chem. Eng. 2015, 7, 113–119, doi:10.1016/j.coche.2014.09.002.
- Kaneko, Y.; Iyi, N.; Kurashima, K.; Matsumoto, T.; Fujita, T.; Kitamura, K. Hexagonal-Structured Polysiloxane Material Prepared by Sol-Gel Reaction of Aminoalkyltrialkoxysilane without Using Surfactants. Mater. 2004, 16, 3417–3423, doi:10.1021/cm0495212.
- Kaneko, Y.; Iyi, N.; Matsumoto, T.; Kitamura, K. Preparation of Higher-Ordered Inorganic-Organic Nanocomposite Composed of Rodlike Cationic Polysiloxane and Polyacrylate. Mater. Chem. 2005, 15, 1572–1575, doi:10.1039/b418579j.
- Kaneko, Y.; Iyi, N.; Matsumoto, T.; Kitamura, K. Synthesis of Rodlike Polysiloxane with Hexagonal Phase by Sol-Gel Reaction of Organotrialkoxysilane Monomer Containing Two Amino Groups. Polymer 2005, 46, 1828–1833, doi:10.1016/j.polymer.2004.12.038.
- Kubo, T.; Koge, S.; Ohshita, J.; Kaneko, Y. Preparation of Imidazolium Salt Type IonicLiquids Containing CyclicSiloxane Frameworks. Lett. 2015, 44, 1362–1364, doi:10.1246/cl.150598.
- Zuo, Y.; Gou, Z.; Li, Z.; Qi, J.; Feng, S. Unexpected Self-Assembly, Photoluminescence Behavior, and Film-Forming Properties of Polysiloxane-Based Imidazolium Ionic Liquids Prepared by One-Pot Thiol–Ene Reaction. New J. Chem. 2017, 41, 14545–14550, doi:10.1039/C7NJ03313C.
- Kaneko, Y.; Iyi, N.; Matsumoto, T.; Fujii, K.; Kurashima, K.; Fujita, T. Synthesis of Ion-Exchangeable Layered Polysiloxane by Sol-Gel Reaction of Aminoalkyltrialkoxysilane: A New Preparation Method for Layered Polysiloxane Materials. Mater. Chem. 2003, 13, 2058–2060, doi:10.1039/b305545k.
- Antonietti, M.; Burger, C.; Effing, J. Mesomorphous Polyelectrolyte-Surfactant Complexes. Mater. 1995, 7, 751–753, doi:10.1002/adma.19950070817.
- Antonietti, M.; Conrad, J.; Thünemann, A. Polyelectrolyte-Surfactant Complexes: A New Type of Solid, Mesomorphous Material. Macromolecules 1994, 27, 6007–6011, doi:10.1021/ma00099a011.
- Sokolov, E.L.; Yeh, F.; Khokhlov, A.; Chu, B. Nanoscale Supramolecular Ordering in Gel-Surfactant Complexes: Sodium Alkyl Sulfates in Poly(Diallyldimethylammonium Chloride). Langmuir 1996, 12, 6229–6234, doi:10.1021/la960274t.
- De Oliveira, V.A.; Tiera, M.J.; Neumann, M.G. Interaction of Cationic Surfactants with Acrylic Acid-Ethyl Methacrylate Copolymers. Langmuir 1996, 12, 607–612, doi:10.1021/la9407774.
- Gainaru, C.; Stacy, E.W.; Bocharova, V.; Gobet, M.; Holt, A.P.; Saito, T.; Greenbaum, S.; Sokolov, A.P. Mechanism of Conductivity Relaxation in Liquid and Polymeric Electrolytes: Direct Link between Conductivity and Diffusivity. Phys. Chem. B 2016, 120, 11074–11083, doi:10.1021/acs.jpcb.6b08567.
- Marangoci, N.; Ardeleanu, R.; Ursu, L.; Ibanescu, C.; Danu, M.; Pinteala, M.; Simionescu, B.C. Polysiloxane Ionic Liquids as Good Solvents for Beta-Cyclodextrin-Polydimethylsiloxane Polyrotaxane Structures. Beilstein J. Org. Chem. 2012, 8, 1610–1618, doi:10.3762/bjoc.8.184.
- Snyder, J.F.; Hutchison, J.C.; Ratner, M.A.; Shriver, D.F. Synthesis of Comb Polysiloxane Polyelectrolytes Containing Oligoether and Perfluoroether Side Chains. Mater. 2003, 15, 4223–4230, doi:10.1021/cm0217396.
- Li, L.; Feng, W.; Welle, A.; Levkin, P.A. UV-Induced Disulfide Formation and Reduction for Dynamic Photopatterning. Chemie 2016, 128, 13969–13973, doi:10.1002/anie.201607276.
- Wang, X.; Bai, L.; Tang, X.; Gao, Y.; Pan, D.; He, X.; Meng, F. Ionic Liquid-Crystalline Network Polymers Formed by Sulfonic Acid-Containing Polysiloxanes and Pyridinium Compounds. Polym. J. 2018, 100, 146–152, doi:10.1016/j.eurpolymj.2018.01.038.
- Mitov, M. Cholesteric Liquid Crystals in Living Matter. Soft Matter 2017, 13, 4176–4209, doi:10.1039/c7sm00384f.
- Chen, Q.; Liang, S.; Shiau, H.S.; Colby, R.H. Linear Viscoelastic and Dielectric Properties of Phosphonium Siloxane Ionomers. ACS Macro Lett. 2013, 2, 970–974, doi:10.1021/mz400476w.
- Liang, S.; Oreilly, M.V.; Choi, U.H.; Shiau, H.S.; Bartels, J.; Chen, Q.; Runt, J.; Winey, K.I.; Colby, R.H. High Ion Content Siloxane Phosphonium Ionomers with Very Low Tg. Macromolecules 2014, 47, 4428–4437, doi:10.1021/ma5001546.
This entry is adapted from the peer-reviewed paper 10.3390/macromol1010002