Fibrillar aggregates and soluble oligomers of both Amyloid-β peptides (Aβs) and hyperphosphorylated Tau proteins (p-Tau-es), as well as a chronic neuroinflammation are the main drivers causing progressive neuronal losses and dementia in Alzheimer's disease (AD). However, the underlying pathogenetic mechanisms are still much disputed. Several endogenous neurotoxic ligands, including Aβs, and/or p-Tau-es activate innate immunity-related danger-sensing/pattern recognition receptors (PPRs) thereby advancing AD's neuroinflammation and progression. The major PRR families involved include scavenger, Toll-like, NOD-like, AIM2-like, RIG-like, and CLEC-2 receptors, plus the calcium-sensing receptor (CaSR). This quite intricate picture stresses the need to identify the pathogenetically topmost Aβ-activated PRR, whose signaling would trigger AD's three main drivers and their intra-brain spread. In theory, the candidate might belong to any PRR family. However, results of preclinical studies using in vitro nontumorigenic human cortical neurons and astrocytes and in vivo AD-model animals have started converging on the CaSR as the pathogenetically upmost PRR candidate. In fact, the CaSR binds both Ca2+ and Aβs and promotes the spread of both Ca2+ dyshomeostasis and AD's three main drivers, causing a progressive neurons' death. Since CaSR's negative allosteric modulators block all these effects, CaSR's candidacy for topmost pathogenetic PRR has assumed a growing therapeutic potential worth clinical testing.
- Alzheimer’s disease
- neuroinflammation
- pattern recognition receptors
- danger-sensing receptors
- inflammasomes
- calcium signaling
1. Introduction
Alzheimer's disease (AD) is the world's most prevalent human dementia. AD is a devastating neurodegenerative illness that slowly yet diffusely kills neurons in cognitively critical areas of the cerebral cortex. Medical advances have both increased the aging fraction of the human population and raised AD's prevalence. Nowadays, AD, particularly in its sporadic (SAD) or late-onset (LOAD) form, affects 35 or more million people, a figure likely to keep rising in next future. Hence, AD has become and will continue being a growingly serious familial, healthcare, and societal burden until the discovery of an effective therapy [1][2]. AD's neuropathology leans on a triad of hallmarks: (i) extracellular congophilic plaques of insoluble fibrillar amyloid-β peptides (fAβs); (ii) intracellular insoluble aggregates (neurofibrillary tangles or NFTs) of hyperphosphorylated Tau proteins (p-Taues); and (iii) a diffuse chronic neuroinflammation [3][4]. But in AD's presymptomatic stage lasting two-to-four decades, other less obvious factors relentlessly drive AD's progression, such as soluble Aβ oligomers (sAβ-os) [5]; soluble p-Tau oligomers (p-Tau-os) [6]; reactive oxygen species (ROS) [7][8]; nitric oxide (NO) and its peroxynitrite derivative (ONOO−) [9]; vascular endothelial growth factor-A (VEGF-A) [10]; and a set of proinflammatory cytokines, chemokines, and other neurotoxic agents [11][12][13]. All such neurotoxins cooperatively cause growing synaptic losses, neural circuits breakdowns, and human neurons and oligodendrocytes deaths, all happening along with a chronically spreading neuroinflammation. The AD neuropathology's clinical counterparts are steadily aggravating losses of memories and cognitive faculties that inexorably lead to patients' dementia and ultimate demise [14][15][16][17][18]. Attempts are under course to identify specific AD's markers that should define the stages of the disease according to scientific criteria [2]. Hitherto, no therapeutic agents, including FDA-approved donepezil, a cholinesterase inhibitor, and memantine, an NMDA receptor antagonist, given singly or in combination, could alter AD's inexorable progression [19].
Continuous advances in medical science and technology have led to an increased debate on the pathophysiology of AD's dementia and to suggest several hypotheses about its pathogenesis (see Table 1).
Table 1. Time-line hypotheses of the causes of SAD/LOAD.
|
Year * |
|
Refs. |
|
1976 |
Cholinergic hypothesis |
[20] |
|
1991 |
Amyloid-β hypothesis |
|
|
1992 |
Calcium dyshomeostasis hypothesis |
[23] |
|
1992 |
Inflammation hypothesis |
[24] |
|
1994 |
Metal ions hypothesis |
[25] |
|
1997 |
Aβ·CaSR activating Ca2+ channels hypothesis |
[26] |
|
2000–2004 |
Neurovascular hypothesis |
|
|
2004 |
Mitochondrial hypothesis |
[30] |
|
2004 |
Glymphatic system hypothesis |
[31] |
|
2009 |
Tau propagation hypothesis |
[32] |
|
2013–2020 |
Aβ·CaSR driving AD progression hypothesis |
[33] |
|
2016 |
Altered gut microbiome hypothesis |
[34] |
|
2018 |
Cellular senescence hypothesis |
[35] |
|
2020 |
Neuroimmunomodulation hypothesis |
[36] |
* The year refers to the first offering of the hypothesis; SAD, sporadic Alzheimer's disease; LOAD, late-onset Alzheimer's disease; CaSR, calcium sensing receptor.
So far, the detailed pathogenesis of SAD/LOAD, which develops into a clinical disease over the course of decades, is still debated due to (i) the structural and functional complexity of human brains; (ii) the lack of specific diagnostic biomarkers useful for an early diagnosis; and (iii) the interplay among several potential risk factors (Table 2).
Table 2. Main factors increasing the risk of SAD/LOAD.
|
Family history |
[37] |
|
Apolipoprotein-ε4 genotype |
[38] |
|
Metabolic syndrome Midlife obesity Hypercholesterolemia Hyperhomocysteinemia Type 2 Diabetes |
|
|
Oxidative stress |
[43] |
|
Midlife hypertension |
[44] |
|
Sleep disorders |
[45] |
|
Oral infections |
[46] |
|
Gut microbiome dysbiosis |
[34] |
|
Human immunodeficiency virus (HIV) |
[47] |
|
Herpes simplex virus type 1 (HSV-1) |
[48] |
The first research endeavors on AD focused on the pathogenetic roles played by Aβ peptides (Aβs) or p-Tau-es, the alternatively primary AD drivers [17][18]. Among a deluge of other hypotheses (Table 1), it was recently set forth that cells of all types in the senescing central nervous system (CNS) might convert the normal ageing process into a neurodegenerative illness [49]. Structural chromatin modifications, irreversible mitotic cell cycle arrest, downregulated expression of lamin B1 and of neurotrophic factors, overexpression and overrelease of proinflammatory agents (e.g., IL-6, etc.) are held to characterize the senescent astrocytes (for further details and references see [50]). However, yet no firmly agreed definition exists about cellular senescence and its triggering mechanisms under the diverse neuropathological conditions. Therefore, it still unclear whether cellular senescence is the cause or outcome of neurodegenerative processes [48]. At any rate, it is remarkable that, whatever be the pathogenetic hypothesis, AD always entails a chronic neuroinflammation. This fact has engendered the "Neuroimmunomodulation Hypothesis of AD", implicating neuroinflammatory phenomena as AD's primary etiological factors [36]. Consequently, the mechanisms underlying AD's neuroinflammation have been attracting an increasing attention [3][4]. Clearly, the clarification of such neuroinflammatory mechanisms and the all-important approaches to counter or mitigate them might hopefully lead to novel and effective treatments of human AD [51].
2. Glia Roles in AD
According to the "Amyloid Hypothesis of AD", the neuropathology starts within the layer II neuronal nests of the temporal lobe lateral entorhinal allocortex and thence via its neuritic projections spreads out toward cognitively crucial upper neocortical areas [52]. The slowly growing load of sAβ42-os and of insoluble Aβ42 fibrils drives the activation of astrocytes and microglia in the brain areas affected. Macroglia (astrocytes and oligodendrocytes) and microglia partake in the innate immune system of the CNS. Microglia are the resident cells responsible for the "immune surveillance". In a "resting" or anti-inflammatory phenotype they upkeep brain homeostasis by secreting anti-inflammatory cytokines, such as TGF-β and IL-10, and neurotrophic factors such as BDNF and GDNF. Thus, homeostatic microglia promote differentiation and survival of neurons, favor learning-dependent synapse formation and plasticity, scavenge neuronal debris, and remove defective neurons by inducing their death [53][54]. Conversely, several noxious stimuli, such as neural tissue damage, exogenous pathogens or endogenous protein aggregates turn on the microglia's activated or proinflammatory phenotype, which gets rid of them through phagocytosis, and secretion of proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β, and of cytotoxic factors such NO and ROS. The roles microglia play in AD are quite complex and heavily debated, with conflicting reports regarding their harmful or shielding impact onto the disease [55]. As shown in transgenic AD-model mice, activated microglia can initially exert beneficial effects on AD pathogenesis by clearing the Aβ-os through phagocytosis [56]. At a later stage, as seen in tissue samples from AD human brains and AD-model animals, the accrual of activated microglia mostly around Aβ plaques, alongside with an increased production of proinflammatory factors, is ultimately noxious to surrounding neurons, thereby advancing disease progression [57].
Astrocytes are the most abundant CNS cell type, from 1.7-fold to 10-fold the neurons' numbers [58] and are the key cell type helping keep CNS homeostasis. Some authors posit that the Aβ-elicited reactive astrogliosis is the leading driver of AD's neuroinflammation [59]. Notably, the proinflammatory activity of the Aβ-driven astrocytes outlasts that of the less plentiful microglia [60][61]. It is important to realize that, once Aβ-activated, astrocytes and microglia reciprocally interact, which further increases the release of a complex set of proinflammatory cytokines, chemokines, and other neurotoxic factors from either cell type [62][63].
A still largely held view posits that in human AD the astrocytes behave as competent phagocytes tasked with cleaning up cellular debris and Aβ42 fibrils becoming overloaded with the latter in the process [63][64][65]. However, cortical nontumorigenic adult human astrocytes (NAHAs) exposed in vitro to fibrillary or soluble Aβ25–35, an Aβ42 proxy, de novo produce, accumulate, and release surpluses of Aβ42-os just like neurons do, while simultaneously decreasing their release of neurotrophic and neuroprotective soluble amyloid precursor protein-α (sAPP-α). Therefore, given their high numbers, the astrocytes can directly contribute to the amyloid brain overload proper of AD [33][66]. In addition, each astrocyte envelops with its mobile processes the so-called tripartite synapses of several neurons [67][68]. Moreover, distinct astrocytes envelop with their processes the synapses of a single neuron. In this way, extended groups of neurons are functionally joined to regulatory astrocytes, while the latter are reciprocally interconnected via gap junctions that allow for the quick diffusion of Ca2+ waves [69]. Regulatory astrocytes also promote the formation and stabilization of their neurons' synapses by mopping up released K+ ions and glutamate. They modulate the neuronal release of neurotransmitters by secreting their own gliotransmitters [70] and via Ca2+ releases and uptakes during their Ca2+ waves [69]. Importantly, astrocytes' processes also envelope cerebral microvessels and affect the local blood flow and oxygen supply to support the functions of their metabolically dependent neurons [70]. An age-related acute or chronic local perfusion deficit and consequent brain tissue hypoxia can cause the accumulation and release of newly produced neurotoxic sAβ-os from both neurons and astrocytes [71]. This engenders vicious feed-forward cycles that stimulate the de novo surplus production and release of further amounts of sAβ-os, p-Tau-os, NO, VEGF-A, proinflammatory cytokines and chemokines, and other neurotoxic agents (Figure 1) [13][15][33][66].
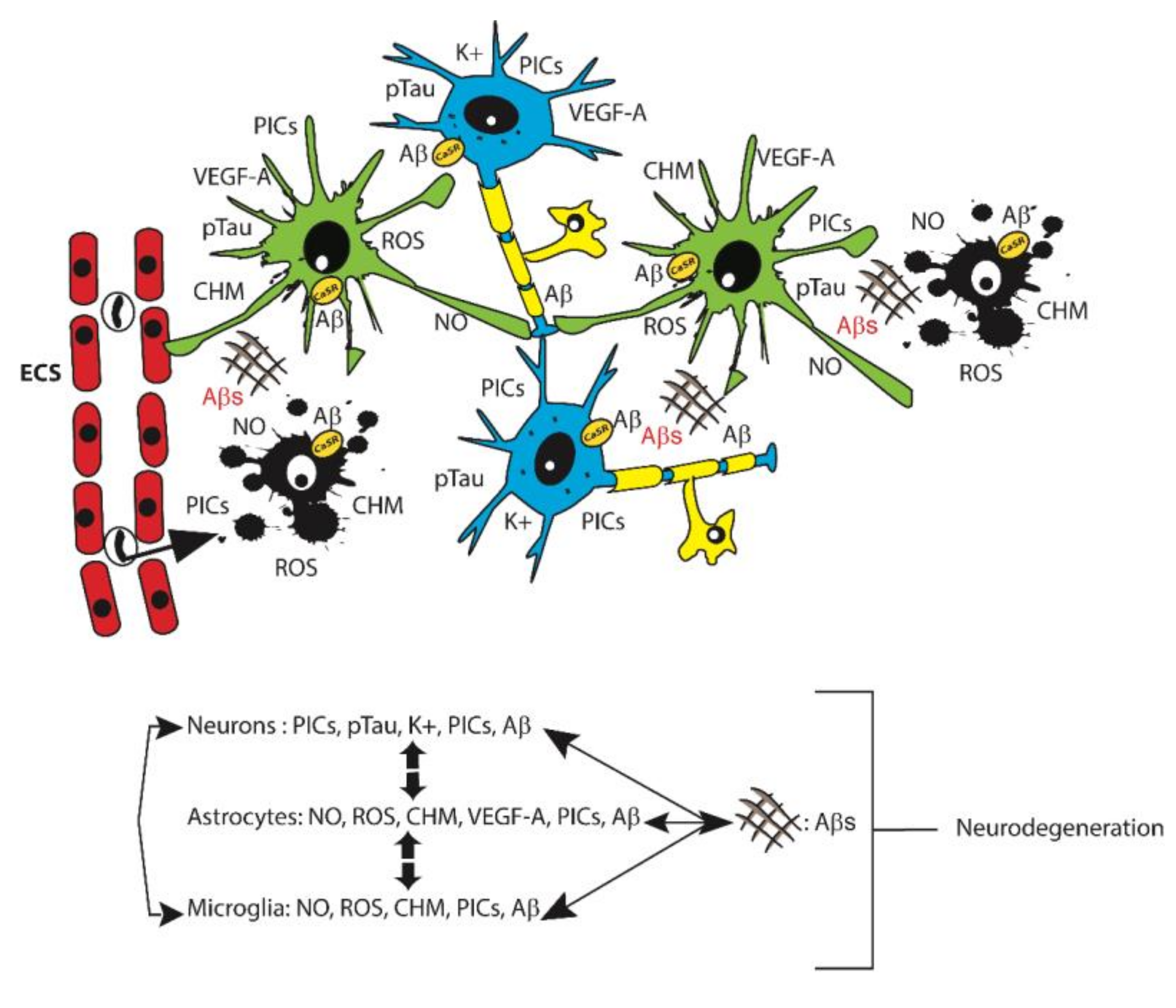
Figure 1. Top: A summary representation of the main cell types and proinflammatory factors each of them releases into the extracellular matrix in the course of AD. Neurons, blue. Astrocytes, green. Oligodendrocytes, yellow. Microglia, black. Endothelial cells (ECs), red. Monocytes, colorless. A black arrow indicates the migration of a monocyte from a capillary lumen into the nervous tissue. Senile plaques, ## Aβs. Most of the abbreviations are as in the text. CHM, chemokines. PICs, proinflammatory cytokines. CaSR, calcium-sensing receptor. Bottom: Schematic diagram of the reciprocal interactions between the three main neural cell types involved in Alzheimer-related neuroinflammation. The bidirectional interaction with amyloid senile plaques is also indicated.
But how can sAβ-os bring about such self-propagating tissue damage? And how might all the involved harming mechanisms be effectively neutralized?
3. Danger-Sensing/Pattern Recognition Receptors (PRRs)
The concept generally accepted in recent years is that the interactions between sAβ-os and a number of multiligand cellular receptors grouped under the denomination of "Pattern Recognition Receptors" (PRRs) may mediate via their intracellular signaling pathways all the noxious effects proper of AD's neuropathology, including the associated neuroinflammation, particularly involving activated microglia and astrocytes [72] and the other CNS cell types too. Therefore, this review will focus on the receptorial interactions of Aβs that feasibly advance AD's progression, particularly about microglia and astrocytes, without neglecting wherever opportune the other CNS cell types.
The evolutionarily highly conserved PRRs are integral parts of the innate immune system. The major PRRs families incorporate scavenger receptors (SRs; e.g., RAGE), Toll-like receptors (TLRs), NOD-like receptors (NLRs), AIM2-like receptors (ALRs), RIG-like receptors (RLRs), CLEC-2 receptors [73], and the calcium-sensing receptor (CaSR) [74]. The sensors of each PRR family pick out a heterogeneous set of specific exogenous pathogen-associated or endogenous damage-associated molecular patterns (PAMPs or DAMPs, respectively) belonging to complex products released from microorganisms or from the different compartments of injured cells or accumulating in the extracellular matrix (ECM). These PAMP•PRR or DAMP•PRR interactions drive the formation of several multicomponent protein signaling platforms, the inflammasomes, evoking the activation of caspase-1 and hence the maturation of proinflammatory cytokines precursors (pro-IL-1β and pro-IL-18), whose release evokes septic or aseptic inflammatory responses. The upshots are PAMPs or DAMPs phagocytosis, synaptic apoptosis (synaptosis), and a caspase-1-dependent inflammatory cell death or pyroptosis [75], and sometimes also the resolution of inflammation. DAMPs contribute to the host's defense by interacting with PRRs, such as RAGE, TLRs, and inflammasomes, and activating the innate immune system [76][77]. However, when dysregulated and/or persistent the same DAMPs•PRRs or PAMPs•PRRs interactions can unphysiologically promote chronic inflammatory responses that cause the development and/or advance the progression of several human inflammatory diseases [76][78][79][80][81][82].
3.1. Scavenger Receptors (SRs)
SRs are cell surface receptors that typically bind multiple ligands and promote the removal of non-self (PAMPs) or altered self (DAMPs) targets. SRs function through mechanisms including endocytosis, phagocytosis, cell adhesion, and intracellular signaling, which lead to the elimination of the degraded or harmful PAMPs or DAMPs. SRs mediate the uptake of fAβs in vitro [83]. SRs are widely distributed in Nature; their nomenclature and classifications have been revised several times [84]. Mammalian SRs include Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) and 1 (TREM1), and Receptor for Advanced Glycation Endproducts (RAGE).
TREM2 is a type I transmembrane cell surface receptor expressed by cells of the myeloid lineage, i.e., monocytes, macrophages, osteoclasts, CNS microglia, and dendritic cells [85][86]. It is a glycoprotein with an immunoglobulin (Ig)-like extracellular domain, a transmembrane domain, and a small cytoplasmic tail. In neural cells, TREM2 has no intracellular signaling system; hence it signals through its transmembrane binding partner, the 12 kDa DNAX activating protein (DAP12 aka TYROBP) that is endowed with an immunoreceptor tyrosine-based activation motif (ITAM) [87]. TREM2′s ligands are multiple, both exogenous PAMPs (from Gram-negative and Gram-positive bacteria, yeasts, and viruses) and endogenous DAMPs (usually polyanionic ligands and phospholipids) [88][89]. TREM2 serves as an anti-inflammatory receptor, negatively regulating the innate immune response via PI3K/NF-κB signaling [89]. Several studies have shown that TREM2 function is crucial for microglial adhesion to and phagocytosis of Aβs, and for neuroinflammation control [90][91][92][93][94]. Interestingly, γ-secretase-dependent intramembranous proteolytic cleavage can shed the soluble (s)TREM2 extracellular domain [95]. sTREM2 may also result from alternative splicing [96]. The cerebrospinal fluid (CSF)'s sTREM2 levels raise with ageing and become further heightened in AD patients, correlating with the CSF levels of total Tau and p-Tau proteins, but not with CSF levels of Aβ42 [97][98].
RAGE belongs to the Immunoglobulin Superfamily and is found on the surface of various immune cells; most of its ligands are mainly secreted by immune cells, including macrophages and dendritic cells; therefore, the major roles played by RAGE relate to inflammation [99]. Neurons, astrocytes, microglia, and vascular cells (pericytes, smooth muscle cells, and endothelial cells) express RAGE in the CNS. Nucleic acids, lipids, and proteins undergo glycations intracellularly within cellular organelles and the cytosol, and within the extracellular matrix (ECM) thus becoming RAGE ligands (AGEs). The expression of both RAGE and its ligands is intense during the embryonic development when they favor the survival of neurons; thereafter, their expression is downregulated and remains at low levels up to old age [100].
Under physiological conditions, anti-glycation defenses efficiently clean off glycated lipids and proteins, while excision repair cuts off glycated nucleotides. AGEs accumulate in aging people and in the course of chronic diseases—such as AD, diabetes, atherosclerosis, arthritis, infections, cancers, and cardiovascular ailments—that are due to various known causes, such as UV light exposure, oxidative stress, malnutrition, epigenetic factors, and to as yet undefined agents. Under such conditions, RAGE and its ligands can be abnormally upregulated in both the CNS and peripheral tissues [101][102]. Accumulating AGEs of various kinds damage cell membranes, cross-link proteins, hamper the function of biological pathways, promote DNA mutations, and curtail mitochondrial ATP production. the intensity of RAGE signals progressively increases as AGEs keep accumulating [103]. As just mentioned, the AGE•RAGE complexes mainly activate the NF-κB intracellular signaling pathway. NF-κB next enters the nucleus and turns on its target genes, including those encoding for several cytokines involved in adaptive or in innate immunity [104][105]. NF-κB targets and downregulates the expression of anti-apoptotic genes, such as those encoding BCL proteins, thereby working against cell survival [106]. Moreover, NF-κB binds glyoxalase (Glo)-1 and curbs its activity inhibiting AGEs production [107]. NF-κB also increases the expression and release of various proinflammatory cytokines, e.g., IL-1β, IL-6, IL-18, and TNF-α from cultured rodent astrocytes. By acting together with NO surpluses, the latter cytokines transform normal astrocytes into reactive astrocytes that promote the onset and/or progression of neurodegenerative disorders [108][109][110]. In addition, RAGE stimulation increases the activities of MAPKs, such as ERK, p38, and JNK, which induce cells to proliferate [111]. As an example of the complexities of RAGE-mediated signaling, let us briefly consider the effects of S100β•RAGE complexes. They did activate three distinct pathways in astrocytes. i.e., the RAGE/Rac-1/Cdc42, the RAGE/ERK/Akt; and/or the RAGE/NF-κB one. The latter next brought about several features proper of reactive astrogliosis, such as hypertrophy, proliferation, and migration. The same astrocytes also acquired a proinflammatory phenotype by expressing IL-1β (the result of inflammasome signaling activation), NO synthase (NOS)-2, and Toll-like receptor (TLR)-2. Finally, the astrocytes also induced an oxygen-glucose deprivation that furthered neurons' death. Altogether, these findings demonstrated that S100β•RAGE complexes concurrently turn on several RAGE-dependent signaling pathways that profoundly change the astrocytes' physiological phenotype into a proinflammatory and proneurodegenerative one [112]. These dysfunctional reactive astrocytes keep oversecreting surpluses of cytokines/chemokines and other proinflammatory mediators, which hinder neuronal glutamate uptake, promote synapses apoptosis (or synaptosis), and advance AD's neuropathology spread, neurons' death, and progressive cognitive deficits [113][114]. Regarding the effects on microglia, the persisting overrelease of proinflammatory cytokines partly prompted by AGE•RAGE signaling hampered microglia's ability to clear the foci of synaptic apoptosis (or synaptosis) occurring without any neuronal death. Synaptosis pruning is mediated on one side by neuronal integrin ανβ3/Tyro3, Axl, and Mer (TAM) receptors plus bridging molecules and on the other side by the microglial PRR C3 receptor. Both these receptor types are necessary for the recognition of focal compartments of synaptic apoptosis exposing phosphatidylserine (PS) and tagged by C1q/C3 complement components [114]. Moreover, AGE•RAGE signaling also undermined microglia's Aβ-clearing activity. Such microglia dysfunctions can advance brain Aβs load and neuroinflammation.
3.2. Toll-Like Receptors (TLRs)
TLRs belong to the PRRs superfamily. Humans have at least ten TLRs, whose expression occurs widely in the brain. Neurons and microglia predominantly express TLRs as compared to astrocytes and oligodendrocytes. TLRs respond to pathogens and cellular stressors (PAMPs) and to DAMPS. In general, TLRs response to pathogens has been more intensely investigated than that to DAMPs. However, regarding the progressive AD-related neuroinflammation the response of TLRs to DAMPs is more relevant. The TLRs ligand-binding extracellular domain is endowed with a variable number of N-terminal leucine-rich repeats (LRRs), which are involved in TLR dimerization [115][116][117]. The signaling of these receptors occurs through their carboxy-terminal intracellular tail that contains a Toll/IL-1 receptor (TIR) homology domain resulting in the recruitment of the cytoplasmic adaptor proteins MyD88 and TOLLIP (Toll interacting protein) and the activation of NF-κB-dependent genes, such as TNF-α, IL-1, IL-6, and IL-8. TLRs are implicated in AD neuropathology by sensing and responding to the presence of different Aβ species. Several studies of postmortem brain tissues from AD patients and transgenic AD-model mice have found augmented expression levels of TLR-2, TLR-4, TLR-5, TLR-7 and TLR-9 as well as of the TLR co-receptor CD14, in microglia localized around senile plaques [117][118][119][120]. The detrimental impact of TLR-2 signaling in AD pathogenesis is due to the polarization of microglia toward a neatly proinflammatory profile (M1). In microglial cells the activation of TLR-2 by fAβs induces the production of proinflammatory mediators, such as NO from inducible NOS-2, TNF-α, IL-1β, and IL-6 [121]. Of note, TLR-2 knockout in APPswe/PS1dE9 mice decreases senile plaques load and mitigates neuronal damage [118][122]. Similarly, TLR-4, when stimulated by fAβs or sAβ-os, induces a strong release of the proinflammatory cytokines IL-1β, IL-6, TNF-α, CCL5, MIP-1α, and MCP-1 [123]. In response to Aβs TLR-2 and TLR-4 might also form complexes with other TLRs (e.g., TLR-4•TLR-6) and with cell surface receptors, e.g., CD14 and SR-B2/CD36, the latter acting as a co-receptor involved in the recognition of fAβs and sAβs. Sheedy et al. [124] showed that the TLR-4•TLR-6 complex and SR-B2/CD36 can harmfully cooperate and direct the activation of the NLRP3 inflammasome signaling pathway in AD-related neuroinflammation. Similarly, the CD14 association with the dimer TLR-2•TLR-4 forms a critical complex that activates microglia and promotes its ability to bind and phagocytose fAβs. Furthermore, the activation of TLR-9 in mice resulted in an increased Aβs uptake and clearance by microglia [125] and in a reactive astrogliosis [126].
3.3. NOD (Nucleotide-Binding Oligomerization Domain) Receptors and (NOD)-Like Leucine-Rich Repeat Receptors (NLRs) Inflammasomes
Inflammasomes are crucial cytoplasmic multimeric protein complexes or platforms that critically regulate the inflammatory responses of innate immunity. Among the most studied ones are the nucleotide binding oligomerization domain receptors 1 and 2 (aka NOD1 and NOD2) [127] and the NLRs (aka nucleotide-binding oligomerization domain [NOD]-like leucine-rich repeat) receptors, which belong to a family of intracellular PRRs initially denoted as cytoplasmic sensors of microbes. Inflammasomes are endowed with intracellular PRR receptors (or sensors) that recognize cell stress-linked DAMPs or pathogen-derived PAMPs. Every type of inflammasome has its own distinct receptors [128][129][130][131]. According to their N-terminus features, NLRs are classified into four subfamilies, i.e., NLRA, NLRB, NLRC, and NLRP. The NLRP family (NLRPs) interacts with the ASC (apoptosis-associated speck-like protein containing a CARD) adaptor protein endowed with an N-terminus PYRIN (aka DAPIN)-CARD domain activating pro-Caspase-1 [132]. In humans, NLRPs comprise 14 members, i.e., NLRP1, NLRP2, NLRP3, and so on. The PYRIN/DAPIN domain crucially controls inflammasome's complex assembly and signal transduction [133]. The ligand specificity of each NLR receptors depends upon their LLRs. Their variable N-terminus domains include caspase-1-binding CARD domain-endowed 4; absent in melanoma 2 (AIM2) protein; and IPAF (ICE protease-activating factor) that effect the activation of distinct biological pathways [134][135][136]. After interacting with PAMPs or DAMPs, PYRIN-PYRIN domains interactions elicit NLRs oligomerization and ASC recruitment [137]. Thereafter, CARD-CARD domains interactions lead to procaspase-1 binding to ASC activate the inflammasome [132]. The formation of complexes between PRR receptors and DAMPs or PAMPs induces the assembly and activates the inflammasomes' signaling that triggers the NF-kB pathway, Caspase-1 activation, and cytokines IL-1β and IL-18 maturation [137][138][139]. Inflammasome activation can induce pyroptosis—an inflammatory form of caspase-1-mediated regulated cell death entailing an initial release through plasmalemmal breaks of the intracellular contents—in various CNS cell types [140][141]. Human neurons, astrocytes, and microglia all display robust NRLP3 inflammasome-associated responses [140]. No investigation concerned inflammasome activation and pyroptosis in human oligodendrocytes until recently when McKenzie et al. [142] observed it in vitro following an exposure to inflammatory stimuli, thus identifying a new mechanism of inflammatory demyelination. Neurons also express the NLRP1 and AIM2 inflammasomes [143], while astrocytes also possess the NLRC4 and NLRP2 ones [138][144] (Table 3).
Table 3. The main inflammasomes expressed in human CNS cell types, in human AD brains and in AD-model animal brains.
|
|
NLRP1 |
NLRP2 |
NLRP3 |
NAIP/ NLRC4 |
AIM2 |
IPAF |
NOD1 |
NOD2 |
NLRP10/ PYNOD |
Refs. # |
|
Neurons |
+ ‡ |
|
+ |
|
+ |
|
|
|
|
|
|
Astrocytes |
|
+ |
+ |
+ |
|
+ |
|
|
|
|
|
Microglia |
+ |
|
+ |
|
|
|
|
|
|
[140] |
|
Oligodendro-cytes |
+ |
|
+ |
|
|
|
|
|
|
[142] |
|
Pericytes |
+ |
+ |
+ |
+ |
|
|
+ |
+ |
|
[146] |
|
Endothelial cells |
+ |
|
+ |
+ |
|
|
+ |
+ |
|
[147] |
|
Postmortem AD human brain |
+ |
|
+ |
+ |
|
+ |
|
|
|
|
|
AD animal models |
+ |
+ |
+ |
+ |
+ |
|
|
|
+ |
#Refs., reference numbers. ‡ +, reported inflammasome in relation to AD neuroinflammation.
The general view holds that PAMPs or DAMPs act as primers inducing the expression and maturation of both the NLR receptors precursors and of Caspase-1. However, it is noteworthy that in the brain inflammasomes preexist as already assembled complexes that do not need the priming step thus giving out faster signals upon activation [164][165]. Therefore, inflammasomes assembly mechanisms may differ from one to another cell type. In response to harmful stimuli neurons, macroglia, and microglia increase the expression of inflammasome proteins, which are key modulators of the innate immune response triggered in AD [165][166][167]. The present understanding of the intricate transactions among so many inflammasomes and their regulatory mechanisms in different pathological conditions is limited: they will need further studies to be clarified.
3.4. C-Type Lectin (Dectin-1, CLEC-2) Receptors (CLRs)
CLRs have diverse functions ranging from embryonic development to immune function. One subgroup of CLRs is the Dendritic cell (DC)-associated C-type lectin-1 (Dectin-1) cluster, comprised of seven receptors including MICL, CLEC-2, CLEC-12B, CLEC-9A, MelLec, Dectin-1 and LOX-1.
The Dectin-1 cluster of receptors has a broad range of ligands and functions. Dectin-1 is an immune-receptor tyrosine-based activation motif (ITAM)-coupled CLR. It senses several DAMPs, recruits the spleen tyrosine kinase (Syk) and lets out proinflammatory signals [168]. Dectin-1 activated the NLRP3 inflammasome for anti-fungal defense and increased IL-1β synthesis via a non-canonical caspase-8 inflammasome [136][169]. After ischemic stroke or spinal cord injury an upregulated Dectin-1 drove neuroinflammation, demyelination, and axons damage through the Syk signaling pathway activation of microglia [170][171]. Conversely, the Dectin-1/Syk pathway inhibition mitigated microglial activation, the brain infarct volume, neurological damage, proinflammatory cytokines production, TNF-α, and NOS-2 expression. Conversely, β-glucan/Dectin-1 signaling induced a beneficial neuroinflammation promoting the regeneration of traumatically severed retinal ganglion cell axons in mice [172]. Dectin-1 is upregulated in human AD tissues [173][174] and AD mouse models [175][176]. In the amyloidogenic 5XFAD transgenic mouse model Dectin1 helped preserve microglia in a homeostatic state because it can evoke TREM2-like intracellular signals [177].
Recent data have led some authors to propose that changes in the gut microbiota and an increased intestinal permeability might be instrumental in AD pathogenesis. Zonulin is a key modulator that regulates intestinal barrier function. Moreover, the activation of C-type lectin-like immune receptor 2 (CLEC-2), a platelet surface receptor may occur in AD. Significant increases in CLEC-2 and Zonulin levels, as assessed via ELISA assays, occurred in 110 MCI and 110 AD patients vs. 110 controls with no dementia. Furthermore, MCI patients had lower CLEC-2 and Zonulin levels than frank AD patients. Further studies will clarify the potential import of these findings in AD [178].
3.5. RIG-I (Retinoid Acid-Inducible Gene-I)-Like Receptors
Human and murine neurons, astrocytes, and microglia constitutively express the retinoic acid-inducible gene I (RIG-I), which is a key cytosolic immune PRR. As a sensor, RIG-I detects 5'-PPP double-stranded RNAs produced by a variety of viruses and intracellular bacteria. Therefore, RIG-I is responsible for mounting a response against infectious agents besides partaking in neuroinflammatory processes linked with neurodegenerative diseases [179][180].
In the course of a bacterial or viral infection, RIG-I expression becomes more intense according to a pathogen and cell type-specific manner. For example, both surface and cytosolic PRR ligands, like bacterial and viral RNA and DNA, effectively increase RIG-I expression, RIG-I-dependent Interferon Regulatory Factor 3 (IRF3) phosphorylation, and subsequent type I Interferon (IFN) production in human microglia [181][182]. Studies on the recognition of Japanese encephalitis virus (JEV) RNA revealed that in JEV-infected brains neurons act via RIG-I as one of the sources of several proinflammatory agents, such as IL-6, IL-12, p70, MCP-1, IP-10, and TNF-α. Conversely, animals with RIG-I knockout neurons had an increased viral load and released lesser amounts of cytokines/chemokines [183]. Interestingly, CoCl2-induced chemical hypoxia increased RIG-I expression and the production of proinflammatory IL-1β, IL-6, and TNF-α via the interaction of Interferon-Β Promoter Stimulator-1 (IPS-1) with TNF receptor-associated factor 6 (TRAF6) and NF-κB pathway activation in cultured human astrocytes [184]. Finally, a study exploring the expression levels of innate immune proteins in human temporal and occipital cortices proved that RIG-1 expression is significantly heightened in the temporal cortex and plasma of patients with mild cognitive impairment (MCI). Moreover, an exposure to the RIG-1 ligand 5'ppp RNA increased the expression of APP and Aβs in primary human astrocytes. These results have revealed a potential implication of RIG-1 in the MCI phase of AD development [180].
3.6. Calcium-Sensing Receptor (CaSR)
A component of Family C G-protein-coupled receptors (GPCRs), the CaSR is a ubiquitously expressed cationic multiligand receptor and at the same time a DAMP-sensing receptor implicated in several inflammatory diseases [74][185][186]. The first cloning of the highly conserved CaSR gene was from rat parotid glands. CaSR acts as a calciostat rapidly sensing changes in systemic extracellular Ca2+ levels ([Ca2+]e) [187]. It is located at the plasmalemma and can signal both thence and, after endocytosis, intracellularly [188]. Besides Ca2+, its hugely bilobed extracellular domain, named Venus flytrap, binds several other agonists such as mono-, di-, and trivalent ions; amino acids (e.g., phenylalanine; tryptophan); and polycationic agonists such as polyamines; aminoglycoside antibiotics; and last but not least Aβs pathological aggregates [13][66][186]. A seven-pass transmembrane domain (the 7TM) joins the Venus flytrap to the intracellular domain (ICD). Ligand binding at the CaSR's Venus flytrap engenders signals that cross the 7TM domain to reach the ICD and induce its binding to G-proteins such heterotrimeric Gi/o, Gq/11, G12/13, and Gs, and low molecular weight factors like Arf6, RhoA, Ras, Rab1 and Rab11a. The intervening 7TM domain plays a critical role in the activation or suppression of agonist-evoked CaSR signaling as it holds specific binding pockets for CaSR positive allosteric modulators (PAMs or calcimimetics) and for CaSR negative allosteric modulators (NAMs or calcilytics) [189][190][191]. The ligand CaSR/G-protein mechanisms drive an intricate set of signaling pathways, conditioned by the nature of the ligand, the cell type considered, and the specific G-protein involved. Such intracellular signals target (i) transcription factors (TFs); (ii) protein kinases (AKT, PKCs, MAPKs); (iii); phospholipases (A2, C, and D); (iv) second messenger up- or downregulation (e.g., cAMP); and (v) Ca2+ influxes via TRPC6-encoded receptor-operated ion channels [192][193][194]. While functioning as a calciostat the CaSR controls systemic Ca2+ homeostasis via the modulation of parathyroid hormone (PTH) and active Vitamin D3 release, which regulate intestinal Ca2+ absorption, skeletal Ca2+ storage, and kidney Ca2+ resorption [193][194].
Several earlier works reported that tissue inflammation can activate CaSR signaling and, on the other hand, that CaSR signaling can activate tissue inflammation. This occurs in deep skin burn wounds [195]; allergen-sensitized airways of mice and of human asthmatic patients [196]; hypertension-induced rat aorta dysfunctional remodeling [197]; LPS-treated rodent lungs [198]; rheumatoid arthritis monocytes [197]; and a variety of diseases of the adipose tissue [199]; kidneys [200]; colon-rectum [201]; and prostate [202]. Of note, CaSR NAM NPS 2143 elicited an effective anti-inflammatory action in asthma [196] and in LPS-evoked pneumonia [198].
Local gradients of [Ca2+]e increases due to cation releases from cells undergoing necrosis following injury or infection activate CaSR's signaling on mature monocytes and/or macrophages. This attracts the phagocytic cells toward the high [Ca2+]e injury sites, a process the chemokine MCP-1/CCR2 signaling aids by increasing the cells' CaSR expression in a series of positive feed-back cycles [203]. Notably, IL-1β and IL-6 too upregulate CaSR expression in parathyroid glands, kidneys, and human astrocytes by acting upon specific response elements of the CASR gene promoters [204]. Besides, CaSR's activation drives Ca2+ release from the ER [205].
Therefore, a heightened [Ca2+]e works as a DAMP that activates the NLRP3 inflammasome via G protein coupled CaSR signaling [206] and via the phosphatidyl inositol/Ca2+ pathway in association with a concurrent fall in cAMP levels in monocytes and macrophages. Thus, CaSR signaling does partake in the induction of inflammation in human cryopyrin-associated periodic syndromes (CAPs) and in mouse models of carrageenan-evoked swelling of foot pads [74][185]. In monocytes of rheumatoid arthritis and of hypertension-evoked aortic remodeling patients [197] [Ca2+]e increases also trigger the NLRP3 inflammasome signaling via CaSR activation [197]. Moreover, in macrophages and monocytes [Ca2+]e surges also activate the macropinocytosis process and the uptake of MDP (a NOD2 ligand) via CaSR-mediated Gα-protein signaling—both events being followed by phosphorylation of the NF-kB p65 subunit or otherwise blocked by the CaSR NAM NPS 2143 or by removing the extracellular Ca2+ [197][207].
As in the other organs, CaSR's expression is ubiquitous in the brain [208], being most intense in hippocampal neurons, astrocytes, microglia, and ependymal cells [209]. Moreover, in the CNS the CaSR crucially modulates nerve cells mitotic activity, prenatal migration, and differentiation; postnatal neurotransmitters release from synapses [208][209][210][211][212]; K+ fluxes [213][214]; and L-amino acid sensing [215].
Importantly, CaSR's expression and its function increases evoking harmful effects under conditions of acute CNS damage that entail a neuroinflammation, such as ischemia/hypoxia/stroke and subarachnoid hemorrhage (which also causes an ischemia/hypoxia through local tissue compression). In adult Kunming mice subjected to a 2-h-long focal cerebral ischemia via carotid ligation followed by a 22-h-long reperfusion, the activation of the JNK/P38 MAPK signaling pathway increased CaSR expression and neurons' apoptosis, both of which were further boosted by administering Gadolinium trichloride (GdCl3), a CaSR PAM [216]. In a mouse model of subarachnoid hemorrhage, GdCl3 worsened the brain edema, the extent of neurodegeneration, and the intensity of neurological deficits, while the administration of NPS 2143, a CaSR NAM, remarkably reduced all such injuries and deficits. Moreover, NPS 2143 countered the GdCl3-evoked increases in NLRP3 inflammasome signaling, caspase-1 activity, IL-1β synthesis, and CaMKII activity and rescued adenylyl cyclase activity and cAMP normal levels. Of note, KN-93, a selective inhibitor of CaMKII, acted by itself as protectively as CaSR NAM NPS 2143 did, indicating its connection with CaSR signaling [217]. Furthermore, traumatic brain injuries too entailed a CaSR's heightened expression and signaling activity that hampered GABABR‘s inhibitory signaling and hence intensified a noxious hyper excitatory activity of the neurons. Mimicking the protective effects of hypothermia, giving CaSR NAM NPS 89,636 did remarkably reduce trauma-evoked brain tissue damage and motor function disability [218]. However, the latter authors did not assess inflammasome expression in their model. In all the acute conditions just mentioned, hypoxia raised [Ca2+]I and boosted CaSR expression thereby increasing BACE1/β-secretase activity, which resulted in the toxic overproduction of Aβ42s. Again, intraventricularly administered CaSR NAM Calhex 231 mitigated such detrimental effects of hypoxia [219].
Studies employing preclinical models of AD in vitro ("in a Petri dish") made of either NAHAs or postnatal HCN-1A neurons isolated from human cerebral cortex fragments showed that exogenous fAβs or sAβs form Aβ•CaSR complexes that are rapidly endocytosed [11][14][33][188]. The thus induced Aβ•CaSR signaling drove (i) a transient CaSR overexpression in vitro [33], which however escalated with time in the hippocampal neurons and astrocytes of 3xTg [220] and of B6C3-Tg (APPswe/PSEN1dE9) AD-model mice [221]; (ii) a decreased proteasome's function promoting the intracellular accumulation of toxic Aβs in human neurons and astrocytes [33]; (iii) a shift of human APP's metabolism along the amyloidogenic processing (AP) leading to the synthesis and release of neurotoxic Aβ42-os surpluses, which deeply downregulated the extracellular shedding of neurotrophic and neuroprotective soluble (s)APP-α [67]; (iv) the hyperphosphorylation of Tau proteins (p-Taues) by raising the glycogen synthase kinase (GSK)-3β activity and their extracellular overrelease via exosomes [189]; (v) the induction and activation of NOS-2 producing toxic NO surpluses [222]; (vi) the overproduction and instant release of vascular endothelial growth factor (VEGF)-A169 from NAHAs, which would cause BBB's dysfunction in vivo [223]; (vii) an upregulated NAHAs' synthesis and release/shedding of proinflammatory IL-6; InterCellular Adhesion Molecule-1 (ICAM-1; both its holoprotein and soluble fragment); Regulated upon Activation, normal T cell Expressed and presumably Secreted (RANTES); and Monocyte Chemotactic Protein (MCP)-2. Importantly, CaSR NAM NPS 2143 totally suppressed all the just mentioned effects evoked by Aβ•CaSR signaling in NAHAs and HCN-1A neurons including the total quelling of IL-6 secretion surges, and the partial yet significant reduction of ICAM-1, RANTES, and MCP-2 releases [13]; (viii) the slow yet progressive death of the human cortical neurons, which was also fully prevented by CaSR NAM NPS 2143; conversely, the NAHAs survived and kept producing and releasing all the above-mentioned detrimental factors unless CaSR NAM NPS 2143 was also added [33]. The beneficial effects of CaSR NAM 2143 and similar agents showed that cortical human astrocytes and neurons Aβ•CaSR signaling could directly advance AD's neuroinflammation and AD's progression, both of which could be effectively stopped by administering CaSR NAMs [11][12][13][14][33][188]. It is also worth mentioning here that the CaSR PAM (calcimimetic) NPS R-568 administration intensified the release of Aβ42-os from the NAHAs further confirming the noxious role of Aβ•CaSR signaling in AD amyloidosis [33].
Interestingly, Feng et al. [221] reported that CaSR NAM NPS 2143 prevented the loss of neuronal dendritic filopodia and synaptic spines, the downregulation of the presynaptic marker synaptotagmine-1, and of the postsynaptic marker PDS 95, and mitigated cognitive deficits induced by Aβ1-42-os in B6C3-Tg (APPswe/PSEN1dE9) AD-model mice and in cultured primary hippocampal neurons.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21239036
References
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015. The global impact of dementia: An analysis of prevalence, incidence, cost and trends. Alzheimer’s Disease International; https://www.alzint.org (Accessed on 21st September 2015)
- Knopman, D.S.; Haeberlein, S.B.; Carrillo, M.C.; Hendrix, J.A.; Kerchner, G.; Margolin, R.; Maruff, P.; Miller, D.S.; Tong, G.; Tome, M.B.; et al. The National Institute on Aging and the Alzheimer's Association Research framework for Alzheimer’s disease: Perspectives from the research roundtable. Alzheimers Dement. 2018, 14, 563–575, doi:10.1016/j.jalz.2018.03.002.
- Labzin, L.I.; Heneka, M.T.; Latz. E. Innate immunity and neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449, doi:10.1146/annurev-med-050715-104343.
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59, doi:10.1038/nri.2016.123.
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid pep–tides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 2015, 12, 3–11, doi:10.1007/s13311-014-0313-y.
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508, doi:10.1001/jamaneurol.2013.5847.
- Della Bianca, V.; Dusi, S.; Bianchini, E.; Dal Pra, I.; Rossi, F. Beta-amyloid activates the O−2 forming NADPH oxidase in microglia, monocytes, and neutrophils: A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol Chem. 1999, 274, 15493–15499.
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367, doi:10.3233/JAD-170543.
- Chiarini, A.; Dal Pra, I.; Menapace, L.; Pacchiana, R.; Whitfield, J.F.; Armato, U. Soluble amyloid β-peptide and myelin basic protein strongly stimulate, alone and in synergism with joint proinflammatory cytokines, the expression of functional nitric oxide synthase-2 in normal adult human astrocytes. Int. J. Mol. Med. 2005, 16, 801–807, doi:10.3892/ijmm.16.5.801.
- Dal Prà, I.; Armato, U.; Chioffi, F.; Pacchiana, R.; Whitfield, J.F.; Chakravarthy, B.; Gui, L.; Chiarini, A. The Aβ peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromol. Med. 2014, 16, 645–657, doi:10.1007/s12017-014-8315-9.
- Dal Prà, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-Sensing Receptors of Human Astrocyte-Neuron Teams: Amyloid-β-Driven Mediators and Therapeutic Targets of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 353–364, doi:10.2174/1570159X12666140828214701.
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-sensing receptors of human neural cells play crucial roles in Alzheimer’s disease. Front. Physiol. 2016, 7, 134, doi:10.3389/fphys.2016.00134.
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes. Cells 2020, 9, 1386, doi:10.3390/cells9061386.
- Dal Prà, I.; Armato, U.; Chiarini, A. Specific interactions of calcium-sensing receptors (CaSRs) with soluble amyloid-β peptides—A study using cultured normofunctioning adult human astrocytes. In Proceedings of the 2nd International Symposium on the Calcium-sensing Receptor, San Diego, CA, USA, 3–4 March 2015; pp. 90–91.
- Dal Prà, I.; Chiarini, A.; Gui, L.; Chakravarthy, B.; Pacchiana, R.; Gardenal, E.; Armato, U. Do astrocytes collaborate with neurons in spreading the “infectious” Aβ and Tau drivers of Alzheimer’s disease? Neuroscientist 2015, 21, 9–29, doi:10.1177/1073858414529828.
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732, doi:10.1016/j.jalz.2016.02.010.
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356, doi:10.1126/science.1072994.
- Braak, H.; Del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain 2015, 138 Pt 10, 2814–2833, doi:10.1093/brain/awv23.
- Calhoun, A.; King, C.; Khoury, R.; Grossberg, G.T. An evaluation of memantine ER + donepezil for the treatment of Alzheimer's disease. Expert Opin. Pharmacother. 2018, 19, 1711–1717, doi:10.1080/14656566.2018.1519022.
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403, doi:10.1016/s0140-6736(76)91936-x.
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498, doi:10.1016/0896-6273(91)90052-2.
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388, doi:10.1016/0165-6147(91)90609-v.
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389, doi:10.1523/jneurosci.12-02-00376.1992.
- McGeer, P.L.; Rogers, J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 1992, 42, 447–449, doi:10.1212/wnl.42.2.447.
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467, doi:10.1126/science.8073293.
- Ye, C.; Ho-Pao, C.L.; Kanazirska, M.; Quinn, S.; Rogers, K.; Seidman, C.E.; Seidman, J.G.; Brown, E.M.; Vassilev, P.M. Amyloid-beta proteins activate Ca(2+)-permeable channels through calcium-sensing receptors. J. Neurosci. Res. 1997, 47, 547–554, doi:10.1002/(sici)1097-4547(19970301)47:5<547::aid-jnr10>3.0.co;2-v.
- Kalaria, R.N. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 321–330, doi:10.1016/s0197-4580(00)00125-1.
- de la Torre, J.C. Alzheimer disease as a vascular disorder: Nosological evidence. Stroke 2002, 33, 1152–1162, doi:10.1161/01.str.0000014421.15948.67.
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360, doi:10.1038/nrn1387.
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20, doi:10.1016/j.mehy.2003.12.045.
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344, doi:10.1016/j.neuron.2004.07.017.
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852, doi:10.1074/jbc.M808759200.
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons—Therapeutic relevance to Alzheimer's disease. Biochim. Biophys. Acta 2013, 1832, 1634–1652, doi:10.1016/j.bbadis.2013.04.020.
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537, doi:10.1038/s41598-017-13601-y.
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216, doi:10.1172/JCI95145.
- Maccioni, R.B.; Navarrete, L.P.; González, A.; González-Canacer, A.; Guzmán-Martínez, L.; Cortés, N. Inflammation: A major target for compounds to control Alzheimer's Disease. J. Alzheimers Dis. 2020, 76, 1199–1213, doi:10.3233/JAD-191014.
- Silverman, J.M.; Raiford, K.; Edland, S.; Fillenbaum, G.; Morris, J.C.; Clark, C.M.; Kukull, W.; Heyman, A. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part VI. Family history assessment: A multicenter study of first-degree relatives of Alzheimer’s disease probands and nondemented spouse controls. Neurology 1994, 44, 1253–1259, doi:10.1212/wnl.44.7.1253.
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118, doi:10.1038/nrneurol.2012.263.
- Razay, G.; Vreugdenhil, A.; Wilcock, G. The Metabolic Syndrome and Alzheimer Disease. Arch. Neurol. 2007, 64, 93–96, doi:10.1001/archneur.64.1.93.
- Dias, H.K.; Brown, C.L.; Polidori, M.C.; Lip, G.Y.; Griffiths, H.R. LDL-lipids from patients with hypercholesterolaemia and Alzheimer’s disease are inflammatory to microvascular endothelial cells: Mitigation by statin intervention. Clin. Sci. 2015, 129, 1195–1206, doi:10.1042/CS20150351.
- Whitmer, R.A.; Gunderson, E.P.; Barrett-Connor, E.; Quesenberry, C.P., Jr.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360, doi:10.1136/bmj.38446.466238.E0.
- Zhuo, J.M.; Wang, H.; Praticò, D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol. Sci. 2011, 32, 562–571, doi:10.1016/j.tips.2011.05.003.
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160, doi:10.1038/s41583-019-0132-6.
- Launer, L.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.; Foley, D.; White, L.R.; Havlik, R.J. Midlife blood pressure and dementia: The Honolulu-Asia aging study. Neurobiol. Aging 2000, 21, 49–55, doi:0.1016/s0197-4580(00)00096-8.
- Osorio, R.S.; Pirraglia, E.; Agüera-Ortiz, L.F.; During, E.H.; Sacks, H.; Ayappa, I.; Walsleben, J.; Mooney, A.; Hussain, A.; Glodzik, L.; et al. Greater risk of Alzheimer’s disease in older adults with insomnia. J. Am. Geriatr. Soc. 2011, 59, 559–562, doi:10.1111/j.1532-5415.2010.03288.x.
- Olsen, I.; Singhrao, S.K. Can oral infection be a risk factor for Alzheimer's disease? J. Oral Mcrobiol. 2015, 7, 29143 doi:10.3402/jom.v7.29143.
- Canet, G.; Dias, C.; Gabelle, A.; Simonin, Y.; Gosselet, F.; Marchi, N.; Makinson, A.; Tuaillon, E.; Van de Perre, P.; Givalois, L.; et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell. Neurosci. 2018, 12, 307, doi:10.3389/fncel.2018.00307.
- Lövheim. H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.G.; Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Demen. 2015, 11, 593–599, doi:10.1016/j.jalz.2014.04.522.
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444, doi:10.1038/s41583-020-0325-z.
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte senescence and Alzheimer’s Disease: A review. Front. Aging Neurosci. 2020, 12,148, doi:10.3389/fnagi.2020.00148.
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397, doi:10.1177/1179573520907397.
- Selkoe, D.J. Biochemistry and molecular biology of amyloid β-protein and the mechanism of Alzheimer’s disease. Handb. Clin. Neurol. 2008, 89, 245–260, doi:10.1016/S0072-9752(07)01223-7.
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The role of microglia and macrophages in CNS homeostasis, autoimmunity, and cancer. J. Immunol. Res. 2017, 2017, 5150678, doi:10.1155/2017/5150678.
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249, doi:10.1172/JCI90606.
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-known targets and new opportunities. Front. Aging Neurosci. 2019, 11, 233, doi:10.3389/fnagi.2019.00233.
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001, 7, 612–618, doi:10.1038/87945.
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167, doi:10.2174/187152710791012071.
- Araque, A.; Navarrete, M. Glial cells in neuronal network function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 2375–2381, doi:10.1098/rstb.2009.0313.
- Medeiros, R.; LaFerla, F.M. Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 2013, 239, 133–138, doi:10.1016/j.expneurol.2012.10.007.
- Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2016, 323, 170–182, doi:10.1016/j.neuroscience.2015.01.007.
- Kato, S.; Gondo, T.; Hoshii, Y.; Takahashi, M.; Yamada, M.; Ishihara. T. Confocal observation of senile plaques in Alzheimer’s disease: Senile plaque morphology and relationship between senile plaques and astrocytes. Pathol. Int. 1998, 48, 332–340, doi:10.1111/j.1440-1827.1998.tb03915.x.
- von Bernhardi, R.; Ramirez, G. Microglia-astrocyte interaction in Alzheimer’s disease: Friends or foes for the nervous system? Biol. Res. 2001, 34, 123–128, doi:10.4067/s0716-97602001000200017.
- Nicoll, J.A.; Weller, R.O. A new role for astrocytes: β-amyloid homeostasis and degradation. Trends Mol. Med. 2003, 9, 281–282, doi:10.1016/s1471-4914(03)00109-6.
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J. Neurosci. 2017, 37, 4023–4031, doi:10.1523/JNEUROSCI.3442-16.2017.
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689, doi:10.1038/ncpneuro0355.
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-Sensing Receptor Antagonist NPS 2143 Restores Amyloid Precursor Protein Physiological Non-Amyloidogenic Processing in Aβ-Exposed Adult Human Astrocytes. Sci. Rep. 2017, 7, 1277, doi:10.1038/s41598-017-01215-3.
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192, doi:10.1523/JNEUROSCI.22-01-00183.2002.
- McIver, S.R.; Faideau, M.; Haydon, P.G. Astrocyte-neuron communications. In: Neural-Immune Interactions in Brain Function and Alcohol-Related Disorders; Cui, C., Grandison, L., Noronha, A., Eds.; Springer: Boston, MA, USA, 2013; pp. 31–64, doi:10.1007/978-1-4614-4729-0_2.
- Antanitus, D.S. A theory of cortical neuron-astrocyte interaction. Neuroscientist 1998, 4, 154–159.
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 2010, 330, 1774, doi:10.1126/science.1197623.
- Kettenmann, H.; Ransom, B.R. (Eds.) Neuroglia, 3rd ed.; Oxford University Press: New York, NY, USA, 2013.
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s Disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427, doi:10.3389/fnmol.2017.00427.
- Kigerl, K.A.; Lai, W.; Rivest, S.; Hart, R.P.; Satoskar, A.R.; Popovich, P.G. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J. Neurochem. 2007, 102, 37–50, doi:10.1111/j.1471-4159.2007.04524.x.
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127, doi:10.1038/nature11588.
- Labbé, K.; Saleh, M. Pyroptosis: A Caspase-1-dependent programmed cell death and a barrier to infection. In The Inflammasomes. Progress in Inflammation Research; Couillin, I., Pétrilli, V., Martinon, F., Eds.; Springer: Basel, Switzerland, 2011; pp. 17–36; doi:10.1007/978-3-0348-0148-5_2.
- Lee, M.S.; Kim, Y.J. Pattern-recognition receptor signaling initiated from extracellular, membrane, and cytoplasmic space. Mol. Cells. 2007, 23, 1–10.
- Thundyil, J.; Lim, K.L. DAMPs and neurodegeneration. Ageing Res Rev. 2015, 24 Pt A, 17–28, doi:10.1016/j.arr.2014.11.003.
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245, doi:10.1074/jbc.R114.619304.
- Santoni, G.; Cardinali, C.; Morelli, M.B.; Santoni, M.; Nabissi, M.; Amantini, C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J. Neuroinflammation 2015, 12, 21, doi:10.1186/s12974-015-0239-2.
- Jin, H.S.; Suh, H.W.; Kim, S.J.; Jo, E.K. Mitochondrial control of innate immunity and inflammation. Immune Netw. 2017, 17, 77–88, doi:10.4110/in.2017.17.2.77.
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J. Histochem. Cytochem. 2018, 66, 213–227, doi:10.1369/0022155417740880.
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112, doi:10.1038/s41577-019-0215-7.
- Verdier, Y.; Penke, B. Binding sites of amyloid β-peptide in cell plasma membrane and implications for Alzheimer's disease. Curr. Protein Pept. Sci. 2004, 5, 19–31, doi:10.2174/1389203043486937.
- Prabhudas, M.; Bowdish, D.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; Means, T.K.; Moestrup, S.K.; et al. Standardizing scavenger receptor nomenclature. J. Immunol. 2014, 192, 1997–2006, doi:10.4049/jimmunol.1490003.
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453, doi:10.1038/nri1106.
- Hickman, S.E.; El Khoury, J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 495–498, doi:10.1016/j.bcp.2013. 11.021.
- Klesney-Tait, J.; Turnbull, I.R.; Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 2006, 7, 1266–1273, doi:10.1038/ni1411. PMID: 17110943.
- Cannon, J.P.; O'Driscoll, M.; Litman, G.W. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 2012, 64, 39–47, doi:10.1007/s00251-011-0562-4.
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599, doi:10.4049/jimmunol.171.2.594.
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 2015, 160, 1061–1071, doi:10.1016/j.cell.2015.01.049..
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657, doi:10.1084/jem.20041611.
- Jiang, T.; Tan, L.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Tan, M.S.; Gu, L.Z.; Wang, H.F.; Ding, Z.Z.; Zhang, Y.D.; et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer's disease. Neuropsychopharmacology 2014, 39, 2949–2962, doi:10.1038/npp.2014.164.
- Kawabori, M.; Kacimi, R.; Kauppinen, T.; Calosing, C.; Kim, J.Y.; Hsieh, C.L.; Nakamura, M.C.; Yenari, M.A. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 2015, 35, 3384–3396, doi:10.1523/JNEUROSCI.2620-14.2015.
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839, doi:10.1002/glia.22966.
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036, doi:10.1074/jbc.M113.517540.
- Schmid, C.D.; Sautkulis, L.N.; Danielson, P.E.; Cooper, J.; Hasel, K.W.; Hilbush, B.S.; Sutcliffe, J.G.; Carson, M.J. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002, 83, 1309–1320, doi:10.1046/j.1471-4159.2002.01243.x.
- Piccio, L.; Deming, Y.; Del-Águila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933, doi:10.1007/s00401-016-1533-5.
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3, doi:10.1186/s13024-016-0071-x.
- Maillard-Lefebvre, H.; Boulanger, E.; Daroux, M.; Gaxatte, C.; Hudson, B.I.; Lambert, M. Soluble receptor for advanced glycation end products: A new biomarker in diagnosis and prognosis of chronic inflammatory diseases. Rheumatology 2009, 48, 1190–1196, doi:10.1093/rheumatology/kep199.
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 2000, 275, 40096–40105, doi:10.1074/jbc.M006993200.
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J Inflam. 2013, 2013, 403460, doi:10.1155/2013/403460.
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.K.; Ramasamy, R.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) and DIAPH1: Implications for vascular and neuroinflammatory dysfunction in disorders of the central nervous system. Neurochem. Int. 2019, 126, 154–164, doi:10.1016/j.neuint.2019.03.012.
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886, doi:10.1007/s00109-005-0688-7.
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta 2000, 1498, 99–111, doi:10.1016/s0167-4889(00)00087-2.
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071, doi:10.1056/NEJM199704103361506.
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 2018, 55, 1082–1096, doi:10.1007/s12035-017-0394-9.
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255, doi:10.2337/db09-0801.
- Gonzalez-Reyes, R.E.; Rubiano, M.G. Astrocyte’s RAGE: More than just a question of mood. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 39–48, doi:10.2174/1871524916999160505105121.
- Ponath, G.; Schettler, C.; Kaestner, F.; Voigt, B.; Wentker, D.; Arolt, V.; Rothermundt, M. Autocrine S100B effects on astrocytes are mediated via RAGE. J. Neuroimmunol. 2007, 184, 214–222, doi:10.1016/j.jneuroim.2006.12.011.
- Wang, Z.; Li, D.D.; Liang, Y.Y.; Wang, D.S.; Cai, N.S. Activation of astrocytes by advanced glycation end products: Cytokines induction and nitric oxide release. Acta Pharmacol. Sin. 2002, 23, 974–980.
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17, doi:10.1186/1479-5876-7-17.
- Villarreal, A.; Seoane, R.; González Torres, A.; Rosciszewski, G.; Angelo, M.F.; Rossi, A.; Barker, P.A.; Ramos, A.J. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: Implications for its role in the propagation of reactive gliosis. J. Neurochem. 2014, 131, 190–205, doi:10.1111/jnc.12790.
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer's disease. Inflammopharmacology 2019, 27, 663–677, doi:10.1007/s10787-019-00580-x.
- Nonaka, S.; Nakanishi, H. Microglial clearance of focal apoptotic synapses. Neurosci. Lett. 2019, 707, 134317, doi:10.1016/j.neulet.2019.134317.
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525, doi:10.1126/science.1085536.
- Crack, P.J.; Bray, P.J. Toll-like receptors in the brain and their potential roles in neuropathology. Immunol. Cell Biol. 2007, 85, 476–480, doi:10.1038/sj.icb.7100103.
- Hopkins, P.A.; Sriskandan, S. Mammalian Toll-like receptors: To immunity and beyond. Clin. Exp. Immunol. 2005, 140, 395–407, doi:10.1111/j.1365-2249.2005.02801.x.
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer's disease. J. Immunol. 2008, 181, 7254–7262, doi:10.4049/jimmunol.181.10.7254.
- Letiembre, M.; Liu, Y.; Walter, S.; Hao, W.; Pfander, T.; Wrede, A.; Schulz-Schaeffer, W.; Fassbender, K. Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiol. Aging 2009, 30, 759–768, doi:10.1016/j.neurobiolaging.2007.08.018.
- Liu, Y.; Walter, S.; Stagi, M.; Cherny, D.; Letiembre, M.; Schulz-Schaeffer, W.; Heine, H.; Penke, B.; Neumann, H.; Fassbender, K. LPS receptor (CD14): A receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 2005, 128, 1778–1789, doi:10.1093/brain/awh531.
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell. Physiol. Biochem. 2007, 20, 947–956, doi:10.1159/000110455.
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107, doi:10.4049/jimmunol.1101121.
- Balducci, C.; Frasca, A.; Zotti, M.; La Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L.; et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav. Immun. 2017, 60, 188–197, doi:10.1016/j.bbi.2016.10.012.
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820, doi:10.1038/ni.2639.
- Tahara, K.; Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain 2006, 129 Pt 11, 3006–3019, doi:10.1093/brain/awl249.
- Tauber, S.C.; Ebert, S.; Weishaupt, J.H.; Reich, A.; Nau, R.; Gerber, J. Stimulation of Toll-like receptor 9 by chronic intraventricular unmethylated cytosine-guanine DNA infusion causes neuroinflammation and impaired spatial memory. J. Neuropathol. Exp. Neurol. 2009, 68, 1116–1124, doi:10.1097/NEN.0b013e3181b7fde5.
- Kufer, T.A.; Sansonetti, P.J. Sensing of bacteria: NOD a lonely job. Curr. Opin. Microbiol. 2007, 10, 62–69, doi:10.1016/j.mib.2006.11.003.
- Ye, Z.; Ting, J.P. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008, 20, 3–9, doi:10.1016/j.coi.2008.01.003.
- Boivin, A.; Pineau, I.; Barrette, B.; Filali, M.; Vallières, N.; Rivest, S.; Lacroix, S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J. Neurosci. 2007, 27, 12565–12576, doi:10.1523/JNEUROSCI.3027-07.2007.
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16, doi:10.1016/j.expneurol.2014.01.001.
- de Rivero Vaccari, J.P.; Minkiewicz, J.; Wang, X.; de Rivero Vaccari, J.C.; German, R.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. Astrogliosis involves activation of retinoic acid-inducible gene-like signaling in the innate immune response after spinal cord injury. Glia 2012, 60, 414–421, doi:10.1002/glia.22275.
- Kufer, T.A.; Sansonetti, P.J. NLR functions beyond pathogen recognition. Nat. Immunol. 2011, 12, 121–128, doi:10.1038/ni.1985. PMID: 21245903.
- Huber, R.G.; Eibl, C.; Fuchs, J.E. Intrinsic flexibility of NLRP pyrin domains is a key factor in their conformational dynamics, fold stability, and dimerization. Protein Sci. 2015, 24, 174–181, doi:10.1002/pro.2601.
- Kufer, T.A.; Kremmer, E.; Banks, D.J.; Philpott, D.J. Role for Erbin in Bacterial Activation of Nod2. Infect. Immun. 2006, 74, 3115-3124, doi:10.1128/IAI.00035-06.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832, doi:10.1016/j.cell.2010.01.040.
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151, doi:10.1111/j.1600-065X.2011.01046.x.
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735, doi:10.1146/annurev-immunol-031210-101405.
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121, doi:10.1002/glia.22499.
- Vladimer, G.I.; Weng, D.; Paquette, S.W.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012, 37, 96–107, doi:10.1016/j.immuni.2012.07.006.
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97, doi:10.1038/nrn3638.
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572, doi:10.1016/j.cub.2016.02.019.
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074, doi:10.1073/pnas.1722041115.
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686, doi:10.1038/cdd.2015.16.
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370, doi:10.1084/jem.20150237.
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: Bringing neurons into the neuroinflammation game. Mol. Neurobiol. 2019, 56, 7741–7753, doi:10.1007/s12035-019-1638-7.
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of pattern recognition receptors and activation of the non-canonical inflammasome pathway in brain pericytes. Brain Behav. Immun. 2017, 64, 220–231, doi:10.1016/j.bbi.2017.04.010.
- Nagyőszi, P.; Nyúl-Tóth, Á.; Fazakas, C.; Wilhelm, I.; Kozma, M.; Molnár, J.; Haskó, J.; Krizbai, I.A. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J. Neurochem. 2015, 135, 551–564, doi:10.1111/jnc.13197.
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247, doi:10.1038/ni.1703.
- Ramaswamy, S.B.; Bhagavan, S.M.; Kaur, H.; Giler, G.E.; Kempuraj, D.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; et al. Glia Maturation Factor in the Pathogenesis of Alzheimer's disease. Open Access J. Neurol. Neurosurg. 2019, 12, 79–82.
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218, doi:10.1038/nature02664.
- Christie, L.A.; Su, J.H.; Tu, C.H.; Dick, M.C.; Zhou, J.; Cotman, C.W. Differential regulation of inhibitors of apoptosis proteins in Alzheimer’s disease brains. Neurobiol. Dis. 2007, 26, 165–173, doi:10.1016/j.nbd.2006.12.017.
- Lesné, S.; Gabriel, C.; Nelson, D.A.; White, E.; Mackenzie, E.T.; Vivien, D.; Buisson, A. Akt-dependent expression of NAIP-1 protects neurons against amyloid-{beta} toxicity. J. Biol. Chem. 2005, 280, 24941–24947, doi:10.1074/jbc.M413495200.
- White, C.S.; Lawrence, C.B.; Brough, D.; Rivers-Auty, J. Inflammasomes as therapeutic targets for Alzheimer's disease. Brain Pathol. 2017, 27, 223–234, doi:10.1111/bpa.12478.
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Tang, S.C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.L.; Thundyil, J.; et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790, doi:10.1038/cddis.2013.326.
- Tan, M.S.; Tan, L.; Jiang, T.; Zhu, X.C.; Wang, H.F.; Jia, C.D.; Yu, J.T. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer's disease. Cell Death Dis. 2014, 5, e1382, doi:10.1038/cddis.2014.348.
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76, doi:10.1038/s41586-018-0761-3.
- Sokolowska, M.; Chen, L.Y.; Liu, Y.; Martinez-Anton, A.; Qi, H.Y.; Logun, C.; Alsaaty, S.; Park, Y.H.; Kastner, D.L.; Chae, J.J.; et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J. Immunol. 2015, 194, 5472–5487, doi:10.4049/jimmunol.1401343.
- Ahmed, M.E.; Iyer, S.; Thangavel, R.; Kempuraj, D.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; Zaheer, A. Co-Localization of Glia Maturation Factor with NLRP3 Inflammasome and Autophagosome Markers in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2017, 60, 1143–1160, doi:10.3233/JAD-170634.
- Couturier, J.; Stancu, I.C.; Schakman, O.; Pierrot, N.; Huaux, F.; Kienlen-Campard, P.; Dewachter, I.; Octave, J.N. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflammation 2016, 13, 20, doi:10.1186/s12974-016-0477-y.
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617, doi:10.1007/s00401-018-01957-y.
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402, doi:10.1038/ni.1864.
- Qu, Y.; Misaghi, S.; Izrael-Tomasevic, A.; Newton, K.; Gilmour, L.L.; Lamkanfi, M.; Louie, S.; Kayagaki, N.; Liu, J.; Kömüves, L.; et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012, 490, 539–542, doi:10.1038/nature11429.
- Marwarha, G.; Schommer, J.; Lund, J.; Schommer, T.; Ghribi, O. Palmitate-induced C/EBP homologous protein activation leads to NF-κB-mediated increase in BACE1 activity and amyloid beta genesis. J. Neurochem. 2018, 144, 761–779, doi:10.1111/jnc.14292.
- Abulafia, D.P.; de Rivero Vaccari, J.P.; Lozano, J.D.; Lotocki, G.; Keane, R.W.; Dietrich, W.D. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 2009, 29, 534–544, doi:10.1038/jcbfm.2008.143.
- de Rivero Vaccari, J.P.; Lotocki, G.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. A molecular platform in neurons regulates inflammation after spinal cord injury. J. Neurosci. 2008, 28, 3404–3414, doi:10.1523/JNEUROSCI.0157-08.2008.
- Lechan, R.M.; Toni, R.; Clark, B.D.; Cannon, J.G.; Shaw, A.R.; Dinarello, C.A.; Reichlin, S. Immunoreactive interleukin-1 beta localization in the rat forebrain. Brain Res. 1990, 514, 135–140, doi:0.1016/0006-8993(90)90445-h.
- Heneka, M.T. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017, 27, 220–222, doi:10.1111/bpa.12483.
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917, doi:10.1038/nm.2749.
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; van der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246–254, doi:10.1038/ni.2222.
- Gensel, J.C.; Wang, Y.; Guan, Z.; Beckwith, K.A.; Braun, K.J.; Wei, P.; McTigue, D.M.; Popovich, P.G. Toll-Like Receptors and Dectin-1, a C-Type Lectin Receptor, Trigger Divergent Functions in CNS Macrophages. J. Neurosci. 2015, 35, 9966–9976, doi:10.1523/JNEUROSCI.0337-15.2015.
- Ye, X.C.; Hao, Q.; Ma, W.J.; Zhao, Q.C.; Wang, W.W.; Yin, H.H.; Zhang, T.; Wang, M.; Zan, K.; Yang, X.X.; et al. Dectin-1/Syk signaling triggers neuroinflammation after ischemic stroke in mice. J. Neuroinflammation 2020, 17, 17, doi:10.1186/s12974-019-1693-z.
- Baldwin, K.T.; Carbajal, K.S.; Segal, B.M.; Giger, R.J. Neuroinflammation triggered by β-glucan/dectin-1 signaling enables CNS axon regeneration. Proc. Natl. Acad. Sci. USA 2015, 112, 2581–2586, doi:10.1073/pnas.1423221112.
- Webster, J.A.; Gibbs, J.R.; Clarke, J.; Ray, M.; Zhang, W.; Holmans, P.; Rohrer, K.; Zhao, A.; Marlowe, L.; Kaleem, M.; et al. Genetic control of human brain transcript expression in Alzheimer disease. Am. J. Hum. Genet. 2009, 84, 445–458, doi:10.1016/j.ajhg.2009.03.011.
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.; Ngu, H.; et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s Disease not evident in mouse models. Cell Rep. 2018, 22, 832–847, doi:10.1016/j.celrep.2017.12.066.
- Wes, P.D.; Easton, A.; Corradi, J.; Barten, D.M.; Devidze, N.; DeCarr, L.B.; Truong, A.; He, A.; Barrezueta, N.X.; Polson, C.; et al. Tau overexpression impacts a neuroinflammation gene expression network perturbed in Alzheimer’s disease. PLoS ONE 2014, 9, e106050, doi:10.1371/journal.pone.0106050.
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31, doi:10.1186/s40478-015-0203-5.
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trümbach, D.; Wurst, W.; et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017, 18, 1186–1198, doi:10.15252/embr.201743922.
- Wang, X.; Liu, G.J.; Gao, Q.; Li, N.; Wang, R.T. C-type lectin-like receptor 2 and zonulin are associated with mild cognitive impairment and Alzheimer's disease. Acta Neurol. Scand. 2020, 141, 250–255, doi:10.1111/ane.13196.
- Patel, J.R.; García-Sastre, A. Activation and regulation of pathogen sensor RIG-I. Cytokine Growth Factor Rev. 2014, 25, 513–523, doi:10.1016/j.cytogfr.2014.08.005.
- de Rivero Vaccari, J.P.; Brand, F.J., 3rd; Sedaghat, C.; Mash, D.C.; Dietrich, W.D.; Keane, R.W. RIG-1 receptor expression in the pathology of Alzheimer’s disease. J. Neuroinflammation 2014, 11, 67, doi:10.1186/1742-2094-11-67.
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Lorière, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I recognizes the 5' Region of Dengue and Zika virus genomes. Cell Rep. 2018, 24, 320–328, doi:10.1016/j.celrep.2018.06.047.
- Johnson, M.B.; Halman, J.R.; Burmeister, A.R.; Currin, S.; Khisamutdinov, E.F.; Afonin, K.A.; Marriott, I. Retinoic acid inducible gene-I mediated detection of bacterial nucleic acids in human microglial cells. J. Neuroinflammation 2020, 17, 139, doi:10.1186/s12974-020-01817.
- Zheng, B.; Wang, X.; Liu, Y.; Li, Y.; Long, S.; Gu, C.; Ye, J.; Xie, S.; Cao, S. Japanese Encephalitis Virus infection induces inflammation of swine testis through RIG-I-NF-ĸB signaling pathway. Vet. Microbiol. 2019, 238, 108430, doi:10.1016/j.vetmic.2019.108430.
- Li, L.; Yang, R.; Feng, M.; Guo, Y.; Wang, Y.; Guo, J.; Lu, X. Rig-I is involved in inflammation through the IPS-1/TRAF6 pathway in astrocytes under chemical hypoxia. Neurosci. Lett. 2018, 672, 46–52, doi:10.1016/j.neulet.2018.02.035.
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329, doi:10.1038/ncomms2339.
- Chiarini, A.; Armato, U.; Dal Prà, I.; Family C G-Protein-Coupled Receptors in Alzheimer’s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282, doi:10.3389/fphar.2019.01282.
- Brown, E.M. The pathophysiology of primary hyperparathyroidism. J. Bone Miner. Res. 2002, 17 (Suppl. 2), N24-29.
- Breitwieser, G.E. The calcium sensing receptor life cycle: Trafficking, cell surface expression, and degradation. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 303–313, doi:10.1016/j.beem.2013.03.003.
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer's Therapy. Front. Neurosci. 2017, 11, 217, doi:10.3389/fnins.2017.00217.
- Leach, K.; Gregory, K.J.; Kufareva, I.; Khajehali, E.; Cook, A.E.; Abagyan, R.; Conigrave, A.D.; Sexton, P.M.; Christopoulos, A. Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res. 2016, 26, 574–592, doi:10.1038/cr.2016.36.
- Nemeth, E.F.; Goodman, W.G. Calcimimetic and calcilytic drugs: Feats, flops, and futures. Calcif. Tissue Int. 2016, 98, 341–358, doi:10.1007/s00223-015-0052-z.
- Chakravarty, B.; Chattopadhyay, N.; Brown, E.M. Signaling through the extracellular calcium-sensing receptor (CaSR). Adv. Exp. Med. Biol. 2012, 740, 103–142, doi:10.1007/978-94-007-2888-2_5.
- Zhang, C.; Miller, C.L.; Brown, E.M.; Yang, J.J. The calcium sensing receptor: From calcium sensing to signaling. Sci. China Life Sci. 2015, 58, 14–27, 10.1007/s11427-014-4779-y.
- Hofer, A.M.; Brown, E.M. Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 530–538, doi:10.1038/nrm1154.
- Klein, G.L.; Castro, S.M.; Garofalo, R.P. The calcium-sensing receptor as a mediator of inflammation. Semin. Cell Dev. Biol. 2016, 49, 52–56, doi:10.1016/j.semcdb.2015.08.006.
- Yarova, P.L.; Stewart, A.L.; Sathish, V.; Britt, R.D., Jr.; Thompson, M.A.P.; Lowe, A.P.; Freeman, M.; Aravamudan, B.; Kita, H.; Brennan, S.C.; et al. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci. Transl. Med. 2015, 7, 284ra60, doi:10.1126/scitranslmed.aaa0282.
- Zhang, X.; Hong, S.; Qi, S.; Liu, W.; Zhang, X.; Shi, Z.; Chen, W.; Zhao, M.; Yin, X. NLRP3 Inflammasome is involved in Calcium-Sensing Receptor-induced aortic remodeling in SHRs. Mediat. Inflamm. 2019, 2019, 6847087, doi:10.1155/2019/6847087.
- Lee, J.W.; Park, H.A.; Kwon, O.K.; Park, J.W.; Lee, G.; Lee, H.J.; Lee, S.J.; Oh, S.R.; Ahn, K.S. NPS 2143, a selective calcium-sensing receptor antagonist inhibits lipopolysaccharide-induced pulmonary inflammation. Mol. Immunol. 2017, 90, 150–157, doi:10.1016/j.molimm.2017.07.012.
- Mattar, P.; Bravo-Sagua, R.; Tobar, N.; Fuentes, C.; Troncoso, R.; Breitwieser, G.; Lavandero, S.; Cifuentes, M. Autophagy mediates calcium-sensing receptor-induced TNFα production in human preadipocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3585–3594, doi:10.1016/j.bbadis.2018.08.020.
- Hu, B.; Tong, F.; Xu, L.; Shen, Z.; Yan, L.; Xu, G.; Shen, R. Role of Calcium Sensing Receptor in streptozotocin-induced diabetic rats exposed to renal ischemia reperfusion injury. Kidney Blood Press. Res. 2018, 43, 276–286, doi:10.1159/000487685.
- Iamartino, L.; Elajnaf, T.; Kallay, E.; Schepelmann, M. Calcium-sensing receptor in colorectal inflammation and cancer: Current insights and future perspectives. World J. Gastroenterol. 2018, 24, 4119–4131, doi:10.3748/wjg.v24.i36.4119.
- Bernichtein, S.; Pigat, N.; Barry Delongchamps, N.; Boutillon, F.; Verkarre, V.; Camparo, P.; Reyes-Gomez, E.; Méjean, A.; Oudard, S.M.; Lepicard, E.M.; et al. Vitamin D3 prevents calcium-induced progression of early-stage prostate tumors by counteracting TRPC6 and Calcium Sensing Receptor upregulation. Cancer Res. 2017, 77, 355–365, doi:10.1158/0008-5472.CAN-16-0687.
- Olszak, I.T.; Poznansky, M.C.; Evans, R.H.; Olson, D.; Kos, C.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J. Clin. Investig. 2000, 105, 1299–1305, doi:10.1172/JCI9799.
- Hendy, G.N.; Canaff, L. Calcium-Sensing Receptor Gene: Regulation of Expression. Front. Physiol. 2016, 7, 394, doi:10.3389/fphys.2016.00394.
- Bagur, R.; Hajnoczky, G. Intracellular Ca(2+) sensing: Its role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788, doi:10.1016/j.molcel.2017.05.028.
- Hendy, G.N.; Canaff, L. Calcium-sensing receptor, proinflammatory cytokines and calcium homeostasis. Semin. Cell. Dev. Biol. 2016, 49, 37–43, doi:10.1016/j.semcdb.2015.11.006.
- Canton, J.; Schlam, D.; Breuer, C.; Gütschow, M.; Glogauer, M.; Grinstein, S. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat. Commun. 2016, 7, 11284, doi:10.1038/ncomms11284.
- Bandyopadhyay, S.; Tfelt-Hansen, J.; Chattopadhyay, N. Diverse roles of extracellular calcium-sensing receptor in the central nervous system. J. Neurosci. Res. 2010, 88, 2073–2082, doi:10.1002/jnr.22391.
- Yano, S.; Brown, E.M.; Chattopadhyay, N. Calcium-sensing receptor in the brain. Cell Calcium 2004, 35, 257–264, doi:10.1016/j.ceca.2003.10.008.
- Chattopadhyay, N.; Ye, C.; Yamaguchi, T.; Nakai, M.; Kifor, O.; Vassilev, P.M.; Nishimura, R.N.; Brown, E.M. The extracellular calcium-sensing receptor is expressed in rat microglia and modulates an outward K+ channel. J. Neurochem. 1999, 72, 1915–1922, doi:10.1046/j.1471-4159.1999.0721915.x.
- Riccardi, D.; Kemp, P.J. The calcium-sensing receptor beyond extracellular calcium homeostasis: Conception, development, adult physiology, and disease. Annu. Rev. Physyiol. 2012, 74, 271–297, doi:10.1146/annurev-physiol-020911-153318.
- Ruat, M.; Traiffort, E. Roles of the calcium sensing receptor in the central nervous system. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 429–442, doi:10.1016/j.beem.2013.03.001.
- Noh, J.S.; Pak, H.J.; Shin, Y.J.; Riew, T.R.; Park, J.H.; Moon, Y.W.; Lee, M.Y. Differential expression of the calcium-sensing receptor in the ischemic and border zones after transient focal cerebral ischemia in rats. J. Chem. Neuroanat. 2015, 66–67, 40–51, doi:10.1016/j.jchemneu.2015.05.001.
- Guo, Y.; Yang, X.; He, J.; Liu, J.; Yang, S.; Dong, H. Important roles of the Ca2+-sensing receptor in vascular health and disease. Life Sci. 2018, 209, 217–227, doi:10.1016/j.lfs.2018.08.016.
- Conigrave, A.D.; Hampson, D.R. Broad-spectrum L-amino acid sensing by class 3 G-protein-coupled receptors. Trends Endocrinol. Metab. 2006, 17, 398–407, doi:10.1016/j.tem.2006.10.012.
- Zhen, Y.; Ding, C.; Sun, J.; Wang, Y.; Li, S.; Dong, L. Activation of the calcium-sensing receptor promotes apoptosis by modulating the JNK/p38 MAPK pathway in focal cerebral ischemia-reperfusion in mice. Am. J. Transl. Res. 2016, 8, 911–921.
- Wang, C.; Jia, Q.; Sun, C.; Jing, C. Calcium sensing receptor contribute to early brain injury through the CaMKII/NLRP3 pathway after subarachnoid hemorrhage in mice. Biochem. Biophys. Res. Commun. 2020, 530, 651–657, doi:10.1016/j.bbrc.2020.07.081.
- Kim, J.Y.; Kim, N.; Yenari, M.A.; Chang, W. Hypothermia and pharmacological regimens that prevent overexpression and overactivity of the extracellular calcium-sensing receptor protect neurons against traumatic brain injury. J. Neurotrauma 2013, 30, 1170–1176, doi:10.1089/neu.2012.2691.
- Bai, S.; Mao, M.; Tian, L.; Yu, Y.; Zeng, J.; Ouyang, K.; Yu, L.; Li, L.; Wang, D.; Deng, X.; et al. Calcium sensing receptor mediated the excessive generation of β-amyloid peptide induced by hypoxia in vivo and in vitro. Biochem. Biophys. Res. Commun. 2015, 459, 568–573, doi:10.1016/j.bbrc.2015.02.141.
- Gardenal, E.; Chiarini, A.; Armato, U.; Dal Prà, I.; Verkhratsky, A.; Rodríguez, J.J. Increased Calcium-Sensing Receptor Immunoreactivity in the Hippocampus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2017, 11, 81, doi:10.3389/fnins.2017.00081.
- Feng, C.; Bao, X.; Shan, L.; Ling, Y.; Ding, Y.; Wang, J.; Cao, Y.; Wang, Q.; Cui, W.; Xu, S. Calcium-Sensing Receptor Mediates β-amyloid-induced synaptic formation impairment and cognitive deficits via regulation of cytosolic Phospholipase A2/Prostaglandin E2 metabolic pathway. Front. Aging Neurosci. 2020, 12, 144, doi:10.3389/fnagi.2020.00144.
- Dal Prà, I.; Chiarini, A.; Nemeth, E.F.; Armato, U.; and Whitfield, J.F. Roles of Ca2+ and the Ca2+-sensing receptor (CaSR) in the expression of inducible NOS (nitric oxide synthase)-2 and its BH4 (tetrahydrobiopterin)-dependent activation in cytokine-stimulated adult human astrocytes. J. Cell. Biochem. 2005, 96, 428–438, doi:10.1002/jcb.20511.
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 129, 120–129, doi:10.1111/jnc.12611.
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 129, 120–129, doi:10.1111/jnc.12611.