Helicobacter pylori infection is a leading cause of gastric cancer, which is the second-most common cancer-related death in the world. The chronic inflammatory environment in the gastric mucosal epithelia during H. pylori infection stimulates intracellular signaling pathways, namely inflammatory signals, which may lead to the promotion and progression of cancer cells.
- Helicobacter pylori
- Gastric Cancer
- inflammatory signals
1. Introduction
Helicobacter pylori is a microaerophilic bacterium that infects the gastric mucosal epithelium through colonization and triggers chronic inflammation [1]. Originally, it was discovered as a curved bacillus in the stomach of patients with gastritis and peptic ulceration by Marshall and Warren [2]. H. pylori produces proteases, vacuolating cytotoxin A (VacA), and certain phospholipases, all of which damage the gastric epithelial cells and disrupt tight junctions, thereby inducing inflammatory changes to gastric mucosa. H. pylori also produces urease that catalyzes the hydrolysis of urea to yield ammonia and carbonic acid, facilitating bacterial colonization by neutralizing gastric acid and providing materials for bacterial protein synthesis [3]. Chronic gastric inflammation caused by the products secreted by H. pylori, which stimulates transcription and synthesis of inflammatory cytokines, especially interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) [4], is considered to play an important role in the development and promotion of gastric cancer, and other organs may see an increased risk of cancer under such inflammatory conditions [5].
Although a persistent inflammatory condition is known to be key to H. pylori-induced gastric carcinogenesis, the molecular mechanisms underlying how H. pylori bacteria directly or indirectly interact with gastric epithelial cells, leading to gastric carcinogenesis, is unclear. In particular, how the inflammatory signals interact with intracellular pathways in gastric epithelial cells, ultimately leading to cell growth and differentiation, remains elusive.
2. H. pylori Infection and Immunity
H. pylori, like other pathogenic bacteria, produces and secretes various toxins outside of cells. Lipopolysaccharide (LPS) is a lipid A and polysaccharide complex comprising the outer membrane of the cell wall that is unique to Gram-negative bacteria. Unlike other secretory toxins, LPS remains on the outer membrane, the outermost layer of bacterial cell wall, and is thus also called an endotoxin. Once LPS binds to receptors on the target cell membrane, it impairs the cell function. LPS is the most potent microbial mediator contributing to the pathogenesis of sepsis and septic shock. When LPS enters the blood stream, it binds to and triggers mononuclear cells to produce pro-inflammatory mediators, including TNF-α and IL-1, which stimulate subsequent reactions in the neutrophil–endothelial cell adhesion and blood coagulation system to activate clotting and help generate micro-thrombi. Recent reports have indicated that chronic inflammation induced by LPS is involved in the progression of several diseases including obesity, type II diabetes, atherosclerosis, neuro-immune disorders, diverse metabolic diseases, and cancers [6][7][8][9][10][11].
LPS derived from H. pylori induces chronic inflammatory injury in gastric mucosa but has shown weaker endotoxic activity than that from Escherichia coli, likely due to the fact that lipid A (glycolipid terminal structure) of H. pylori-derived LPS does not have a typical β-1,6-diglucosamine skeleton, which is well-recognized in LPS derived from other Gram-negative bacteria, like E. coli [12][13]. Therefore, it is the H. pylori- but not E. coli-derived LPS that attenuates the cytotoxicity of mononuclear cells (MNCs) against gastric cancer cells. H. pylori LPS also downregulates the perforin production in CD56+ natural killer cells (NK cells) co-cultured with cancer cells, while E. coli LPS does not. In addition, it has also been reported that the milieu of H. pylori LPS is accompanied by the proliferation of regulatory IL-10-producing NK cells, which negatively regulates the cytotoxic activity of the gastric epithelia in the H. pylori-infected hosts. In contrast, NK cells in the milieu of E. coli LPS do not tend to produce IL-10 [14][15].
Thus, H. pylori LPS is considered to diminish the propagation and cytotoxic activity of NK cells that constitute the first line of anti-cancer immune defense, thereby leading to chronic infection of the gastric epithelia without severe cell damage [15]. In addition, the TNF-α mRNA expression is significantly lower in MNCs stimulated with H. pylori than in those stimulated with E. coli under the same conditions [16]. Although H. pylori LPS is reported to induce a high production of neutrophil-recruiting CXC chemokines similar to E. coli LPS, it is less potent for inducing pro-inflammatory cytokines in MNCs and a weak inducer of the CC chemokine RANTES (regulated on activation, normal T-cell expressed and secreted: RANTES) [17]. Importantly, while E. coli LPS is a potent inducer of TNF-α, IL-1β, IL-6, and IL-8, which function through the activation of nuclear factor-κB (NF-κB), H. pylori LPS inhibits most of these factors, including IL-1β, IL-6, and IL-8, and selectively upregulates the production of IL-18 and IL-12 [12]. (Figure 1). Specifically, H. pylori LPS binds to transmembrane Toll-like receptor 4 (TLR4) on the surface of MNCs and activates the TLR4- and toll/IL-1 receptor domain-containing adaptor inducing interferon-β (TRIF)-dependent pathways, which convert pro-IL-1β and pro-IL-18 to IL-1β and IL-18 [18][19]. Of note, IL-1β binds to its receptors on naïve T-cells and promotes Th1 and Th17 differentiation, leading to pro-inflammatory activities, whereas IL-18 counteracts IL-1β by preventing the Th17 response, Treg differentiation, and immune tolerance persistence, thereby balancing the control of infection [20][21][22]. The Th17 response is also reported to be driven by the H. pylori-secreted peptidyl prolyl cis, trans-isomerase, which promotes the pro-inflammatory low cytotoxic gastric tumor-infiltrating lymphocytes response, matrix degradation, and pro-angiogenic pathways, ultimately leading to the promotion of gastric cancer [23].
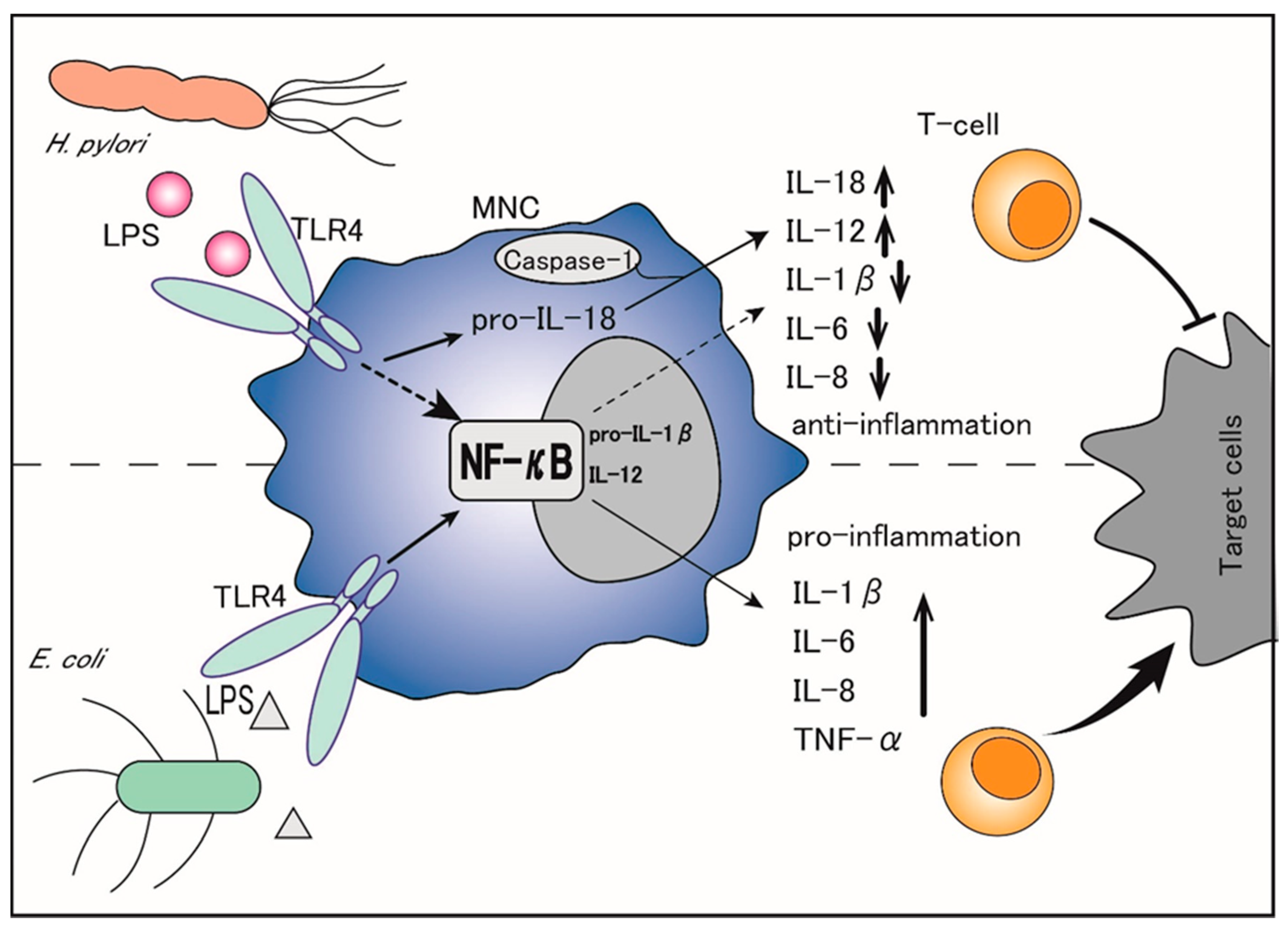
Thus, by selectively upregulating IL-18 but suppressing IL-1β, H. pylori LPS not only inhibits the immune inflammatory response but also promotes gastric cancer cells’ escape from immune surveillance, which facilitates gastric cancer initiation and progression.
Using an in vivo guinea pig model, the difference in the role of H. pylori-derived components such as glycine acid extract (GE), urease subunit A (UreA), CagA, and LPS has been discussed [24]. H. pylori infection promoted the infiltration of inflammatory cells in the gastric mucosa, which caused increased oxidative stress and cell apoptosis. Originally, the elimination of damaged cells by apoptosis has the effect of preventing deleterious inflammation and neoplasia. However, the upregulation of cell regeneration induced by GE, UreA, and CagA may be a risk factor for neoplasia. In contrast, H. pylori LPS had the effect of downregulating cell regeneration, which was shown to promote chronic inflammatory processes [24]. An in vitro study also showed that H. pylori LPS—but not CagA or other surface components of bacteria—inhibited cell migration and proliferation [25]. H. pylori LPS promoted chronic inflammation and inhibited the anti-bacterial response of immune cells, which may constitute the niche for neoplastic processes.
There are several reports on other negative immunomodulatory properties of H. pylori infection. The ability of mononuclear cells to present antigens to lymphocytes, lymphocyte proliferation, and phagocytic activity of the infiltrated macrophages were all adversely affected by H. pylori infection [26][27][28]. By showing these negative immunomodulatory mechanisms, H. pylori not only inhibits the inflammatory immune response and promotes their survival as well as long-term infection, but in turn also increases the risk of mutations of gastric epithelial cells. It helps the immune escape of cancer cells and contributes to the progression of cancer.
This entry is adapted from the peer-reviewed paper 10.3390/jcm9113699
References
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740.
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315.
- Mobley, H.L. The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment Pharmacol. Ther. 1996, 10 (Suppl. 1), 57–64.
- Sugimoto, M.; Furuta, T.; Yamaoka, Y. Influence of inflammatory cytokine polymorphisms on eradication rates of Helicobacter pylori. J. Gastroenterol. Hepatol. 2009, 24, 1725–1732.
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081.
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90.
- Faraj, T.A.; McLaughlin, C.L.; Erridge, C. Host defenses against metabolic endotoxaemia and their impact on lipopolysaccharide detection. Int. Rev. Immunol. 2017, 36, 125–144.
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiol. Bethesda 2016, 31, 283–293.
- Benson, S.; Engler, H.; Schedlowski, M.; Elsenbruch, S. Experimental endotoxemia as a model to study neuroimmune mechanisms in human visceral pain. Ann. N. Y. Acad. Sci. 2012, 1262, 108–117.
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204.
- Waldner, M.J.; Neurath, M.F. Mechanisms of Immune Signaling in Colitis-Associated Cancer. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 6–16.
- Shimoyama, A.; Saeki, A.; Tanimura, N.; Tsutsui, H.; Miyake, K.; Suda, Y.; Fujimoto, Y.; Fukase, K. Chemical synthesis of Helicobacter pylori lipopolysaccharide partial structures and their selective proinflammatory responses. Chemistry 2011, 17, 14464–14474.
- Fujimoto, Y.; Shimoyama, A.; Saeki, A.; Kitayama, N.; Kasamatsu, C.; Tsutsui, H.; Fukase, K. Innate immunomodulation by lipophilic termini of lipopolysaccharide; synthesis of lipid As from Porphyromonas gingivalis and other bacteria and their immunomodulative responses. Mol. Biosyst. 2013, 9, 987–996.
- Rudnicka, K.; Matusiak, A.; Miszczyk, E.; Rudnicka, W.; Tenderenda, M.; Chmiela, M. Immunophenotype of peripheral blood natural killer cells and IL-10 serum levels in relation to Helicobacter pylori status. Apmis 2013, 121, 806–813.
- Rudnicka, K.; Miszczyk, E.; Matusiak, A.; Walencka, M.; Moran, A.P.; Rudnicka, W.; Chmiela, M. Helicobacter pylori-driven modulation of NK cell expansion, intracellular cytokine expression and cytotoxic activity. Innate Immun. 2015, 21, 127–139.
- Ahmadzadeh, E.; Zarkesh-Esfahani, H.; Roghanian, R.; Akbar, F.N. Comparison of Helicobacter pylori and Escherichia coli in induction of TNF-alpha mRNA from human peripheral blood mononuclear cells. Indian J. Med. Microbiol. 2010, 28, 233–237.
- Innocenti, M.; Svennerholm, A.M.; Quiding-Jarbrink, M. Helicobacter pylori lipopolysaccharides preferentially induce CXC chemokine production in human monocytes. Infect. Immun. 2001, 69, 3800–3808.
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001, 19, 423–474.
- Kanneganti, T.D.; Lamkanfi, M.; Kim, Y.G.; Chen, G.; Park, J.H.; Franchi, L.; Vandenabeele, P.; Núñez, G. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 2007, 26, 433–443.
- Hitzler, I.; Sayi, A.; Kohler, E.; Engler, D.B.; Koch, K.N.; Hardt, W.D.; Müller, A. Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1β and IL-18. J. Immunol. 2012, 188, 3594–3602.
- Koch, K.N.; Hartung, M.L.; Urban, S.; Kyburz, A.; Bahlmann, A.S.; Lind, J.; Backert, S.; Taube, C.; Müller, A. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J. Clin. Investig. 2015, 125, 3297–3302.
- Shimada, M.; Ando, T.; Peek, R.M.; Watanabe, O.; Ishiguro, K.; Maeda, O.; Ishikawa, D.; Hasegawa, M.; Ina, K.; Ohmiya, N.; et al. Helicobacter pylori infection upregulates interleukin-18 production from gastric epithelial cells. Eur. J. Gastroenterol. Hepatol. 2008, 20, 1144–1150.
- Amedei, A.; Munari, F.; Bella, C.D.; Niccolai, E.; Benagiano, M.; Bencini, L.; Cianchi, F.; Farsi, M.; Emmi, G.; Zanotti, G.; et al. Helicobacter pylori secreted peptidyl prolyl cis, trans-isomerase drives Th17 inflammation in gastric adenocarcinoma. Intern. Emerg. Med. 2014, 9, 303–309.
- Gonciarz, W.; Krupa, A.; Hinc, K.; Obuchowski, M.; Moran, A.P.; Gajewski, A.; Chmiela, M. The effect of Helicobacter pylori infection and different H. pylori components on the proliferation and apoptosis of gastric epithelial cells and fibroblasts. PLoS ONE 2019, 14, e0220636.
- Mnich, E.; Kowalewicz-Kulbat, M.; Sicińska, P.; Hinc, K.; Obuchowski, M.; Gajewski, A.; Moran, A.P.; Chmiela, M. Impact of Helicobacter pylori on the healing process of the gastric barrier. World J. Gastroenterol. 2016, 22, 7536–7558.
- Mnich, E.; Gajewski, A.; Rudnicka, K.; Gonciarz, W.; Stawerski, P.; Hinc, K.; Obuchowski, M.; Chmiela, M. Immunoregulation of antigen presenting and secretory functions of monocytic cells by Helicobacter pylori antigens in relation to impairment of lymphocyte expansion. Acta Biochim. Pol. 2015, 62, 641–650.
- Schwartz, J.T.; Allen, L.A. Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J. Leukoc. Biol. 2006, 79, 1214–1225.
- Paziak-Domańska, B.; Chmiela, M.; Jarosińska, A.; Rudnicka, W. Potential role of CagA in the inhibition of T cell reactivity in Helicobacter pylori infections. Cell Immunol. 2000, 202, 136–139.