Translational research has revolutionized how we develop new treatments for cancer patients. The change from an organ-centric concept guiding treatment choice towards deep molecular analysis, driving a personalized approach, is one of the most important advances of modern oncology. Several tools such as next generation sequencing and RNA sequencing have greatly improved the capacity to detect predictive and prognostic molecular alterations. Detection of gene mutations, amplifications, and fusions has therefore altered the history of several diseases in both a localized and metastatic setting. This shift in perspective, in which attention is focused on the specific molecular alterations of the tumor, has opened the door to personalized treatment.
- precision medicine
- personalized medicine
- transla
1. The Revolution of Tumor Treatment: from Tumor Site to Molecular Alterations
During the last few years, cancer patient treatment has been completely revolutionized as several molecular alterations have been identified as drivers of cancer development and progression [1]. This biomarker-based approach has historically proceeded from basic science to validation in the clinic. Thus, to guide treatment choice, attention has gradually turned to biomarkers rather than the anatomic site of origin of a given tumor [2]. Due to this change, large groups of patients whose tumors bear a particular molecular feature have been successfully treated with a single targeted drug. The first example of molecular-based medicine for cancer patients was the use of endocrine therapy in luminal breast cancer [3]. However, the point probably marking the revolution towards a precision medicine approach was the approval of imatinib in patients with chronic myeloid leukemia bearing the t (9,22) translocation that creates a BCR-ABL fusion kinase [4].
The identification of druggable molecular alterations marked a new era in oncology [5]. The most important results were achieved in breast [6][7][8][9] and lung cancer [10][11][12] and melanoma [13]. In breast cancer, apart from identifying hormonal receptors as therapeutic agents, other pinpointed biomarkers such as PIK3CA (Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform) or ERBB2 (human epidermal growth factor receptor 2) mutations have completely altered the therapeutic approach in luminal breast cancer patients [14][15][16]. Moreover, the detection of HER2 amplification as a driver has also contributed immensely to locating another important subgroup of patients who definitely benefit from anti-HER2 inhibition in all clinical settings [17][18][19]. A fundamental shift was also observed in patients diagnosed with non-small cell lung cancer (NSCLC). The identification of EGFR (Epidermal Growth Factor Receptor) mutations [20] and EML4-ALK (echinoderm microtubule-associated protein-like 4- Anaplastic lymphoma kinase) translocation [21]
Pinpointing specific molecular characteristics (such as microsatellite instability across all solid tumors), somatic or germline mutations, (such as BRCA, BRAF or ERBB2 HER2) [22][23][24][25], or gene fusions (FGFR and NTRK) [26][27] could guide treatment choice in this field, improving patient outcomes independently of tumor location. To properly apply these molecular-based treatments, it is ultimately necessary to distinguish which patient groups will probably benefit or not from this type of therapy by identifying specific biomarkers predictive of response or resistance [28].
Although genomics seems to be a really relevant start point to plan a precision approach for cancer patients, it is clear that molecular phenotype measurements and characterization are a needed arm to the understanding of tumors to improve the precision medicine approach. In some cases, the use of proteomic could also help when several molecular alterations are detectable, making it difficult to identify the most relevant driver to be targeted [29]. Moreover, genetic mutations do not always result in the predicted change of the corresponding protein, and many other factors contribute to tumor behavior, such as protein modifications, metabolism, and the microbiome [30][31]. Another important field to try to improve a precision medicine approach is metabolomic [32]. The importance of it is not only for the identification of targetable biomarkers but also for the identification of pharmacological phenotype able to understand the mechanisms of inter-patient variability in response to drug therapy [33]. Metabolomics can also help in evaluating drug resistance and disease relapse [34][35], leading to opportunities for the development of novel therapeutic strategies [32].
Identifying those molecular features has led to a clinical strategy in which testing upfront or after the first-line failure may provide certain novel therapeutic opportunities. In umbrella trials, patients diagnosed with the same type of solid tumors are treated according to their molecular features, while in basket trials, patients diagnosed with different types of solid tumors with a common driver molecular alteration are selected and treated with a specific inhibitor [36]. As an example, a multisite study (including >1000 patients with non-small cell lung cancer) showed that matching was associated with longer survival than was seen in patients without genotype-directed treatment. Likewise, two meta-analyses [37][38] in 70,000 patients reported that trials with a personalized strategy led to a higher proportion of responding patients and longer progression-free and overall survival than trials with unselected patients. Standout results were obtained in patients diagnosed with tumors harboring ERBB2 mutations or NTRK or FGFR fusions (Figure 1).
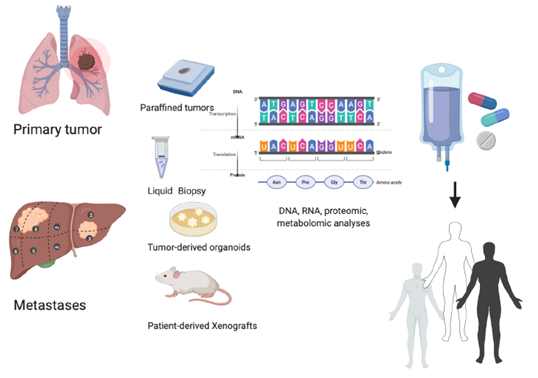
Figure 1. Personalized treatment: an integrated precision approach. How to personalize cancer treatment from the molecular evaluation of primary tumor or metastases evaluating liquid biopsy, tumor-derived organoids, and tumor-derived xenografts.
In the MOSCATO 01 trial, cancer treatment was planned according to genomic analyses. A potentially actionable molecular alteration was identified in 411 of 843 patients. A total of 199 patients were treated with a targeted agent matched to a genomic alteration. The Progression-Free survival 2/1 (PFS2/1) ratio was >1.3 in about one-third of the patients. Although objective responses were observed in only 11% of patients, progression-free survival with this molecularly guided strategy was longer than obtained by the previous line of therapy in one-third of 194 patients. This study suggested that high-throughput genomics could improve outcomes in molecularly selected patients when treated accordingly [39].
2. Precision Molecular Oncology: Understanding the Role of New Drivers with Novel Drugs
Several potential targets for novel drugs have been identified using high-throughput technologies, and several compounds have recently been approved or are under investigation.
One of the most prevalent molecular alterations in solid tumors is PIK3CA mutations. The PI3K/AKT/mTOR (The phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of the rapamycin (mTOR))pathway is an intracellular signaling pathway implicated in cell proliferation. This pathway can be activated at several points, but PIK3CA mutations and PTEN(Phosphatase and tensin homolog) function loss are the most frequent detectable molecular alterations [40]. For this reason, several basket trials have been conducted or are still ongoing to assess the role of PIK3CA inhibitors in several solid tumors. In luminal breast cancer, the incidence of PIK3CA mutations is about 30% in primary tumors and metastases. In a phase III trial enrolling PIK3CA mutant luminal breast cancer patients progressing to hormonal treatment, adding alpelisib, a specific p110α PIK3CA inhibitor, to fulvestrant significantly improved antitumor activity (response rate 26.6% vs. 12.8%) and progression-free survival (11 vs. 5.7 months) over a combination of placebo plus fulvestrant, leading to its regulatory approval. These results confirm that PIK3CA mutations could be drivers in metastatic luminal breast cancer and suggest the importance of detecting this molecular alteration in selected patients. Other more specific novels PI3K inhibitors are currently under clinical development (Table 1).
Table 1. Novel targeted agents against PIK3CA1, FGFR2, and NTRK3 molecular alterations under development for solid tumors in phase I trials.
|
Class of inhibitors |
Novel targeted agents under development for solid tumors in phase I trials |
||
|
Compound name |
Mechanism of action |
Phase |
|
|
PI3K inhibitors |
GDC0077 [41] |
Potent PI3K alpha inhibitor |
Ib |
|
MEN1611 (PA799)[42] |
PI3K alpha inhibitor |
Ib |
|
|
AMG319 [43] |
AMG319 is a PI3Kδ inhibitor. Preclinically, target inhibition abrogates Treg-mediated immunosuppression, augmenting CD8+ T-cell antitumor activity |
IIa |
|
|
CH5132799 [44] |
Oral pan-PI3 kinase inhibitor |
Ia/b |
|
|
FGFR inhibitors |
AZD4547 [45] |
A potent and selective inhibitor of FGFR 1, 2, and 3 |
I |
|
NVP-BGJ398 [46] |
Oral, selective, ATP-competitive inhibitor of FGFR1, 2, and 3 |
I |
|
|
E-7090 [47] |
An oral and selective inhibitor of FGFR1, 2, and 3 |
I |
|
|
LY2874455 [48] |
Inhibitor of FGFR 1, 2, 3, and 4 |
I |
|
|
TAS-120 [49] |
Potent and highly specific against wildtype FGFR1–4 as well as against some FGFR2 kinase domain mutations |
I |
|
|
BLU-554 [50] |
A potent and selective inhibitor of FGFR4 |
I |
|
|
H3B-6527 [51] |
Selective and covalent inhibitor of FGFR4 |
I |
|
|
FGF-401 [52] |
Potent and selective, reversible-covalent small-molecule inhibitor of FGFR4 |
I |
|
|
NTRK inhibitor |
LOXO-195 [53] |
A selective inhibitor of TRK |
I |
|
TSR-011 [54] |
Dual ALK4 and TRK inhibitor |
I |
|
|
DS-6051b [55] |
Inhibitor with high affinity for ROS1 5 and TRK |
I |
|
1 PIK3CA= Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha; FGFR 2= Fibroblast Growth Factor Receptor; NTRK 3= Neurotrophic tropomyosin receptor kinas; ALK 4= Anaplastic lymphoma kinase; ROS1 5= c-ros oncogene 1.
Activation of fibroblast growth factor receptor (FGFR) pathways also plays a major role. FGFRs are made up of four highly conserved transmembrane tyrosine kinases receptors (FGFR1–4) and FGFR5, which lacks an intracellular kinase domain. All these receptors are activated by fibroblast growth factor (FGF), participating in cell survival and proliferation. Deregulation of the FGF signaling axis is implicated in oncogenesis, tumor progression, and resistance to anticancer therapy across several solid tumors, with a prevalence below 10%. Multiple trials studying diverse solid tumors have proposed the aberrant FGFR signaling pathway as a potential therapeutic target, and several inhibitors are under development. FGFR alterations may consist of amplification, mutations, and gene-fusion, yet the specific role of each one and their contribution in predicting drug response is not clear [56]. In early clinical trials, promising results were reported in patients who show FGFR2 amplification in gastric cancer and patients with FGFR2 and FGFR3 translocations in cholangiocarcinoma and urothelial cancers, respectively. Disappointingly, only modest levels of clinical activity have been reported for patients with other aberrations, such as FGFR1 amplification7 or FGFR2 mutation, in advanced-stage endometrial cancer [57]. Urothelial cancer is one of the tumors that most harbors FGFR alterations. In particular, the luminal I subtype is characterized by a lack of benefit from immunotherapy and a higher rate of mutations in the gene encoding for FGFR, detectable in about 20% of all urothelial cancers [58]. In a phase II study [59] treating patients diagnosed with locally advanced and unresectable or metastatic urothelial carcinoma with FGFR alterations, the potent FGFR1–4 tyrosine inhibitor erdafitinib demonstrated antitumor activity, achieving objective responses in 40% of previously treated patients. Interestingly, patients who had undergone previous immunotherapy also achieved the same benefit. FGFR2 activation by point mutations and FGFR1–3 amplification or overexpression are also observed in subsets of patients with advanced cholangiocarcinoma. The most common alterations are chromosomal fusions consisting of FGFR2 exons 1 to 17, encoding the intact extracellular and kinase domains, fused in-frame to a 3′ partner that possesses a protein dimerization domain. Multiple FGFR-selective inhibitors are being tested in clinical trials in cholangiocarcinoma patients harboring FGFR pathway alterations (Table 1).
BRAF mutations are major oncogenic drivers in 7% of solid tumors [60]. BRAF inhibition has led to significant improvement in melanoma. In colorectal cancer, BRAF mutation has been associated with specific clinicopathological features as well as a very poor prognosis. BRAF mutations are more prevalent in proximal colon tumors, poorer differentiation, mucinous histology, and older females [61]. However, several clinical trials testing BRAF inhibitors as single agents in BRAF-mutant colon cancer patients saw the response in less than 5%, mostly of short duration. This lack of efficacy was attributed to feedback EGFR activation resulting in MAPK (mitogen-activated protein kinase) signaling pathway reactivation. Based on this hypothesis, the combination of BRAF/MEK inhibitors plus monoclonal antibodies against EGFR has been explored. In a phase III trial, 665 metastatic colorectal patients with BRAF-V600E mutation who had experienced treatment failure with one or two prior regimens were recruited to receive binimetinib, encorafenib, and cetuximab versus irinotecan-based chemotherapy. This triplet therapy was well tolerated with a manageable safety profile. Moreover, it showed a better response rate than chemotherapy (26% vs. 2%) as well as significantly improved overall survival (OS) for those receiving the triple targeted agent combination (9 vs. 5.4 months), becoming a new standard of care for previously treated BRAF V600E–mutant metastatic colorectal cancer patients and underlining the need to uncover BRAF mutations in all advanced colon cancer patients [62].
Recently, NTRK fusions have also been detected as a possible target [63]. The neurotrophic tyrosine receptor kinases (NTRK) plays a key role in nervous system development. These are three receptors, TrkA, TrkB, and TrkC, with a tyrosine kinase intracellular domain able to activate signaling pathways like MAPK, PI3K/AKT, and PLCγ/PKC (phospholipase-Cγ and protein kinase C signal pathways) resulting in enhanced migration, cell differentiation, synapse formation, and proliferation. In NTRK fusions, the 3′-region is fused with a 5′ region of a fusion partner, resulting in a chimeric receptor protein with dimerization, and thus, uncontrolled activation of the TRK kinase domain leads to chimeric proteins with constitutively activated or overexpressed kinase function conferring dominant oncogenic potential. These oncogenic fusions can be either intra or interchromosomal. All three NTRK genes are potentially affected. These alterations are considered rare, being present in about 0–25%, with the highest occurrence in secretory breast cancer, mammary analog secretory cancer (MASC) of salivary glands, congenital mesoblastic nephroma and infantile fibrosarcoma thyroid cancers, spitzoid melanomas, and gastrointestinal stromal tumors (GIST) [63]. The presence of NTRK fusion seems to indicate early alterations in solid tumors, so the use of specific inhibitors would supposedly improve clinical outcomes. Larotrectinib, a selective TRK inhibitor, was tested in adults and children whose tumors harbored an NTRK 1-2-3 fusion. The overall response rate was 75%. Other NTRK inhibitors are currently under investigation, and some have shown clinical activity in patients resistant to first-generation inhibitors due to the development of secondary mutations (Table 1 and Figure 2).
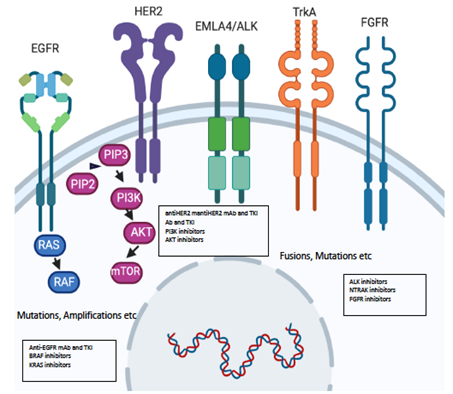
Figure 2. Molecular alterations driving precision oncology.
This entry is adapted from the peer-reviewed paper 10.3390/cancers12041009
References
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365, doi:10.1038/s41571-018-0002-6.
- Lambert, A.W.; Pattabiraman, D.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691, doi:10.1016/j.cell.2016.11.037.
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66.
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49, doi:10.1186/s12943-018-0780-6.
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599, doi:10.1016/j.cell.2016.12.015.
- Ignatiadis, M.; Sotiriou, C. Luminal breast cancer: From biology to treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506, doi:10.1038/nrclinonc.2013.124.
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940, doi:10.1056/NEJMoa1813904.
- Swain, S.; Baselga, J.; Kim, S.-B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.-M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734, doi:10.1056/NEJMoa1413513.
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35.
- Mok, T.S.; Kim, S.W.; Wu, Y.L.; Nakagawa, K.; Yang, J.J.; Ahn, M.J.; Wang, J.; Yang, J.C.H.; Lu, Y.; Atagi, S.; et al. Gefitinib Plus Chemotherapy Versus Chemotherapy in Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer Resistant to First-Line Gefitinib (IMPRESS): Overall Survival and Biomarker Analyses. J. Clin. Oncol. 2017, 35, 4027–4034, doi:10.1200/JCO.2017.73.9250.
- Solomon, B.; Kim, N.-W.; Mekhail, T.; Paolini, J.; Usari, T.; Reisman, A.; Wilner, K.D.; Tursi, J.; Mok, T.S.; Wu, Y.-L.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177, doi:10.1056/nejmoa1408440.
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as fi rst-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Artic. Lancet Oncol. 2012, 13, 239–285.
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Sileni, V.C.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636, doi:10.1056/NEJMoa1904059.
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O'Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819.
- Hyman, D.M.; Piha-Paul, S.; Won, H.; Rodon, J.; Saura, C.; Shapiro, G.I.; Juric, D.; Quinn, D.I.; Moreno, V.; Doger, B.; et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018, 554, 189–194, doi:10.1038/nature25475.
- Smyth, L.M.; Piha-Paul, S.A.; Won, H.H.; Schram, A.M.; Saura, C.; Loi, S.; Lu, J.; Shapiro, G.I.; Juric, D.; Mayer, I.A.; et al. Efficacy and Determinants of Response to HER Kinase Inhibition in HER2-Mutant Metastatic Breast Cancer. Cancer Discov. 2019, 10, 198–213, doi:10.1158/2159-8290.CD-19-0966.
- Krop, I.E.; Kim, S.-B.; González-Martín, A.; Lorusso, P.M.; Ferrero, J.-M.; Smitt, M.; Yu, R.; Leung, A.C.F.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice for pretreated HER2-positive advanced breast cancer (TH3RESA): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 689–699, doi:10.1016/s1470-2045(14)70178-0.
- Doi, T.; Shitara, K.; Naito, Y.; Shimomura, A.; Fujiwara, Y.; Yonemori, K.; Shimizu, C.; Shimoi, T.; Kuboki, Y.; Matsubara, N.; et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody–drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: A phase 1 dose-escalation study. Lancet Oncol. 2017, 18, 1512–1522, doi:10.1016/s1470-2045(17)30604-6.
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791, doi:10.1056/NEJMoa1209124.
- Paez, J.G. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500, doi:10.1126/science.1099314.
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566, doi:10.1038/nature05945.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Adashek, J.J.; Lorusso, P.M.; Hong, D.S.; Kurzrock, R. Phase I trials as valid therapeutic options for patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 773–778, doi:10.1038/s41571-019-0262-9.
- Gonzalez-Angulo, A.M.; Timms, K.M.; Liu, S.; Chen, H.; Litton, J.K.; Potter, J.; Lanchbury, J.S.; Stemke-Hale, K.; Hennessy, B.T.; Arun, B.K.; et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res. 2011, 17, 1082–1089.
- Alsop, K.; Fereday, S.; Meldrum, C.; DeFazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.; Stewart, C.; et al. BRCA Mutation Frequency and Patterns of Treatment Response in BRCA Mutation–Positive Women with Ovarian Cancer: A Report From the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663, doi:10.1200/jco.2011.39.8545.
- Dienstmann, R.; Rodon, J.; Prat, A.; Perez-Garcia, J.; Adamo, B.; Felip, E.; Cortés, J.; Iafrate, A.J.; Nuciforo, P.; Tabernero, J. Genomic aberrations in the FGFR pathway: Opportunities for targeted therapies in solid tumors. Ann. Oncol. 2014, 25, 552–563, doi:10.1093/annonc/mdt419.
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739, doi:10.1056/NEJMoa1714448.
- Kimmelman, J.; Tannock, I. The paradox of precision medicine. Nat. Rev. Clin. Oncol. 2018, 15, 341–342, doi:10.1038/s41571-018-0016-0.
- Rodriguez, H.; Pennington, S.R. Revolutionizing Precision Oncology through Collaborative Proteogenomics and Data Sharing. Cell 2018, 173, 535–539.
- Mertins, P.; Mani D.R.; Ruggles K.V.; Gillette M.A.; Clauser K.R.; Wang, P.; Wang, X.; Qiao J.W.; Cao, S. Petralia, F.; et al. Proteogenomics connects somatic mutations to signaling in breast cancer. Nature 2016, 534, 55–62.
- Puchades-Carrasco, L.; Lucena, A.P. Metabolomics Applications in Precision Medicine: An Oncological Perspective. Curr. Top. Med. Chem. 2017, 17, 2740–2751, doi:10.2174/1568026617666170707120034.
- Clayton, T.A.; Lindon, J.C.; Cloarec, O.; Antti, H.; Charuel, C.; Hanton, G.; Provost, J.P.; Le Net, J.L.; Baker, D.; Walley, R.J.; et al. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature 2006, 440, 1073–1077.
- Gambardella, V.; Gimeno-Valiente, F.; Tarazona, N.; Martínez-Ciarpaglini, C.; Roda, D.; Fleitas, T.; Tolosa, P.; Cejalvo, J.M.; Huerta, M.; Roselló, S.; et al. NRF2 through RPS6 Activation Is Related to Anti-HER2 Drug Resistance in HER2-Amplified Gastric Cancer. Clin. Cancer Res. 2018, 25, 1639–1649, doi:10.1158/1078-0432.ccr-18-2421.
- Rojo de Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cáncer, Cancer Cell 2019, 34, 21–43.
- Garralda, E.; Dienstmann, R.; Piris-Giménez, A.; Braña, I.; Rodon, J.; Tabernero, J. New clinical trial designs in the era of precision medicine. Mol. Oncol. 2019, 13, 549–557, doi:10.1002/1878-0261.12465.
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J. Clin. Oncol. 2015, 33, 3817–3825, doi:10.1200/jco.2015.61.5997.
- Jardim, D.L.; Schwaederle, M.; Wei, C.; Lee, J.J.; Hong, D.S.; Eggermont, A.M.; Schilsky, R.L.; Mendelsohn, J.; Lazar, V.; Kurzrock, R. Impact of a Biomarker-Based Strategy on Oncology Drug Development: A Meta-Analysis of Clinical Trials Leading to FDA Approval. J. Natl. Cancer Inst. 2015, 107, doi:10.1093/jnci/djv253.
- Massard, C.; Michiels, S.; Ferté, C.; Le Deley, M.-C.; Lacroix, L.; Hollebecque, A.; Verlingue, L.; Ileana, E.; Rosellini, S.; Ammari, S.; et al. High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer Discov. 2017, 7, 586–595, doi:10.1158/2159-8290.cd-16-1396.
- Goncalves, M.D.; Hopkins, B.D.; Cantley, L.C. Phosphatidylinositol 3-Kinase, Growth Disorders, and Cancer. N. Engl. J. Med. 2018, 379, 2052–2062, doi:10.1056/nejmra1704560.
- Hong, R.; Edgar, K.; Song, K.; Steven, S.; Young, A.; Hamilton, P.; Arrazate, A.; De La Cruz, C.; Chan, C.; Pang, J.; et al. Abstract PD4-14: GDC-0077 is a selective PI3Kalpha inhibitor that demonstrates robust efficacy in PIK3CA mutant breast cancer models as a single agent and in combination with standard of care therapies. Cancer Res. 2018, 78, PD4-14.
- Merlino, G.; Fiascarelli, A.; Bigioni, M.; Bressan, A.; Irrissuto, C.; Pellacani, A.; Scaltriti, M.; Binaschi, M. Abstract 2160: MEN1611, a novel α-selective PI3K inhibitor in solid tumors. Tumor Biol. 2018, 78, 2160, doi:10.1158/1538-7445.am2018-2160.
- Ottensmeier, C.H.; Jones, T.; Sacco, J.J.; McCaul, J.; Brennan, P.; Paterson, C.; Schache, A.; Shaw, R.J.; Singh, R.P.; Davies, J.H.; et al. A randomised, double-blind, placebo-controlled phase IIa trial of AMG319 given orally as neoadjuvant therapy in patients with human papillomavirus (HPV) positive and negative head and neck squamous cell carcinoma (HNSCC). J. Clin. Oncol. 2018, 36, 6068, doi:10.1200/jco.2018.36.15_suppl.6068.
- Blagden, S.; Olmin, A.; Josephs, D.; Stavraka, C.; Zivi, A.; Pinato, D.J.; Anthoney, A.; Decordova, S.; Swales, K.; Riisnaes, R.; et al. Correction: First-in-human study of CH5132799, an oral class I PI3K inhibitor, studying toxicity, pharmacokinetics, and pharmacodynamics, in patients with metastatic cancer. Clin. Cancer Res. 2015, 21, 660.
- Paik, P.K.; Shen, R.; Berger, M.F.; Ferry, D.; Soria, J.-C.; Mathewson, A.; Rooney, C.; Smith, N.R.; Cullberg, M.; Kilgour, E.; et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients with Advanced Squamous Cell Lung Cancers. Clin. Cancer Res. 2017, 23, 5366–5373, doi:10.1158/1078-0432.CCR-17-0645.
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.-P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821, doi:10.1158/2159-8290.CD-18-0229.
- Koyama, T.; Shimizu, T.; Iwasa, S.; Fujiwara, Y.; Kondo, S.; Kitano, S.; Yonemori, K.; Shimomura, A.; Iizumi, S.; Sasaki, T.; et al. Abstract B160: First-in-human phase 1 study of E7090, a novel selective inhibitor of FGFRs, in patients with advanced solid tumors. Mol. Cancer Ther. 2018, 17, B160, doi:10.1158/1535-7163.targ-17-b160.
- Michael, M.; Bang, Y.-J.; Park, Y.S.; Kang, Y.-K.; Kim, T.M.; Hamid, O.; Thornton, N.; Tate, S.C.; Raddad, E.; Tie, J. A Phase 1 Study of LY2874455, an Oral Selective pan-FGFR Inhibitor, in Patients with Advanced Cancer. Target. Oncol. 2017, 12, 463–474, doi:10.1007/s11523-017-0502-9.
- Goyal, L.; Shi, L.; Liu, L.Y.; De La Cruz, F.F.; Lennerz, J.K.; Raghavan, S.; Leschiner, I.; Elagina, L.; Siravegna, G.; Ng, R.; et al. TAS-120 Overcomes Resistance to ATP-Competitive FGFR Inhibitors in Patients with FGFR2 Fusion-Positive Intrahepatic Cholangiocarcinoma. Cancer Discov. 2019, 9, 1064–1079, doi:10.1158/2159-8290.CD-19-0182.
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707, doi:10.1158/2159-8290.cd-19-0555.
- Mercade, T.M.; Moreno, V.; John, B.; Morris, J.C.; Sawyer, M.B.; Yong, W.P.; Gutierrez, M.; Karasic, T.B.; Sangro, B.; Sheng-Shun, Y.; et al. A phase I study of H3B-6527 in hepatocellular carcinoma (HCC) or intrahepatic cholangiocarcinoma (ICC) patients (pts). J. Clin. Oncol. 2019, 37, 4095, doi:10.1200/jco.2019.37.15_suppl.4095.
- Weiss, A.; Adler, F.; Buhles, A.; Stamm, C.; Fairhurst, R.A.; Kiffe, M.; Sterker, D.; Centeleghe, M.; Wartmann, M.; Kinyamu-Akunda, J.; et al. FGF401, A First-In-Class Highly Selective and Potent FGFR4 Inhibitor for the Treatment of FGF19-Driven Hepatocellular Cancer. Mol. Cancer Ther. 2019, 18, 2194–2206, doi:10.1158/1535-7163.mct-18-1291.
- Hyman, D.; Kummar, S.; Farago, A.; Geoerger, B.; Mau-Sorensen, M.; Taylor, M.; Garralda, E.; Nagasubramanian, R.; Natheson, M. Abstract CT127: Phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi). Cancer Res. 2019, 79, CT127.
- Lin, C.-C.; Arkenau, H.-T.; Lu, S.; Sachdev, J.; Carpeño, J.D.C.; Mita, M.; Dziadziuszko, R.; Su, W.-C.; Bobilev, D.; Hughes, L.; et al. A phase 1, open-label, dose-escalation trial of oral TSR-011 in patients with advanced solid tumours and lymphomas. Br. J. Cancer 2019, 121, 131–138, doi:10.1038/s41416-019-0503-9.
- Papadopoulos, K.P.; Gandhi, L.; Jänne, P.A.; Ou, S.-H.I.; Shaw, A.; Goldberg, T.R.; Greenberg, J.; Gu, X.; Tachibana, M.; Senaldi, G.; et al. First-in-human study of DS-6051b in patients (pts) with advanced solid tumors (AST) conducted in the US. J. Clin. Oncol. 2018, 36, 2514, doi:10.1200/jco.2018.36.15_suppl.2514.
- Dieci, M.; Arnedos, M.; André, F.; Soria, J.-C. Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discov. 2013, 3, 264–279, doi:10.1158/2159-8290.cd-12-0362.
- Babina, I.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332, doi:10.1038/nrc.2017.8.
- Siefker-Radtke, A.; Curti, B. Immunotherapy in metastatic urothelial carcinoma: Focus on immune checkpoint inhibition. Nat. Rev. Urol. 2017, 15, 112–124, doi:10.1038/nrurol.2017.190.
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.F.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348, doi:10.1056/nejmoa1817323.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954, doi:10.1038/nature00766.
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954, doi:10.1038/nature00766.
- Yaeger, R.; Chatila, W. K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A. et al. Clinicla sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 2019, 33, 125–136.
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643, doi:10.1056/NEJMoa1908075.
- Märkl, B.; Hirschbühl, K.; Dhillon, C. NTRK-Fusions—A new kid on the block. Pathol. Res. Pract. 2019, 215, 152572.