Endoglin is a class I, single-membrane spanning receptor with an apparent molecular weight of 95 kDa containing a short cytoplasmic and a modular extracellular domain. This domain contains attachment sites for N- and O-dependent glycosylation and ligand binding residues.
- Endoglin
- CD105
- TGF-β-signaling
1. Structural and Functional Aspects of Endoglin
The homo-dimeric protein is stabilized by multiple intermolecular disulfide bridges [1,2]. In case of Endoglin, defective N-glycosylation interferes with membrane localization similar to the transforming growth factor-β type II receptor (TβRII), as well as it impacts exosomal targeting of Endoglin [1,3,4]. Direct binding of Endoglin to TGF-β is assumed to only occur in complex with the signaling receptors TGF-β type I and type II [2,5,6]. The Endoglin C-terminus is a substrate for TGF-β-receptors, leading to serine/threonine phosphorylation, which regulates the interaction of Endoglin with those receptors [7]. In addition, the C-terminal domain is tyrosine phosphorylated by Src kinase impacting Endoglin’s trafficking and endothelial responses [8]. In contrast to the signaling receptors, Endoglin possesses only a short intracellular domain with no intrinsic kinase activity. Nevertheless, this domain represents a hub for interaction with TGF-β/bone morphogenetic protein (BMP)-receptors, integrins and proteins regulating migration and trafficking such as zyxin, zyxin-related protein (ZRP)-1, β-arrestin2, and the GAIP C-terminus-interacting protein (GIPC) [9,10,11,12,13,14]. The interaction with GIPC affects in addition to Smads (see below) also PI3K/Akt and ERK1/2 activation (Figure 1) [12,14].
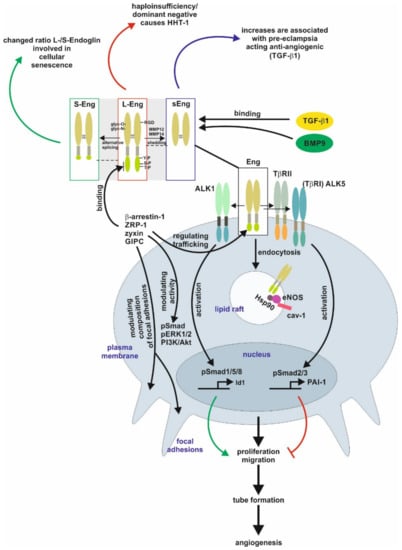
Figure 1. Endoglin biology in endothelial cells. (central part) Endoglin (Eng) is a component of a receptor complex in endothelial cells comprising the type I receptor dimers activin receptor-like kinase 5 (ALK5) or ALK1, and type II receptor dimers binding ligands, e.g., transforming growth factor-β1 (TGF-β1) and bone morphogenetic protein 9 (BMP9), of the TGF-β-superfamily. Interaction with these receptors either activates Smad1/5/8 or Smad2/3 signaling (phosphorylation) to regulate corresponding target genes, e.g., inhibitor of differentiation 1 (Id1) and plasminogen activator inhibitor-1 (PAI-1), and finally, cellular responses including proliferation and migration to balance the activation and resolution phase of angiogenesis. Localization of Eng in caveolin-1 (cav-1) positive lipid rafts mediates interaction of heat shock protein 90 (Hsp90) with endothelial NO-synthase (eNOS) regulating its function. (upper part) Beside the long variant of Eng (L-Eng), a C-terminally truncated, shorter splice variant (S-Eng) and a shedded form, mediated by matrix metalloproteinase (MMP)-14, comprising only the extracellular domain (sEng) has been identified. All three variants of Eng are involved individually in cellular (senescence) and pathological conditions (e.g., HHT-1, pre-eclampsia), respectively. The extracellular domain of Eng is N- and O-glycosylated and human Eng contains an integrin binding motif (RGD) motif. The L-Eng C-terminal domain is a substrate for TGF-β receptors leading to serine/threonine phosphorylation. Tyrosine phosphorylation is mediated by Src. L-Eng interacts with its cytoplasmic domain with several intracellular proteins regulating receptor trafficking, phosphorylation/activity of kinases, and focal adhesion amongst others. The functional difference of S-Eng compared to L-Eng most likely arises from the missing binding- and phosphorylation-sites in its C-terminus. Abbreviations used are: GIPC, GAIP C-terminus-interacting protein; HHT-1, hereditary hemorrhagic telangiectasia 1; Smad, small mothers against decapentaplegic; pERK1/2, phosphorylated extracellular signal-regulated kinase 1/2; PI3K/Akt, phosphatidylinositol 3-kinase; TβRI/II, transforming growth factor-β1 type I or II receptor; ZRP-1, zyxin-related protein-1.
Beside the “regular” Endoglin variant (L-Endoglin), a shorter splice variant of Endoglin (S-Endoglin) has been identified [15,16,17]. This splice variant has a shortened C-terminus, which is missing phosphorylation and protein interaction sites, resulting in functional differences compared to L-Endoglin (see below). Upon ligand binding, TβRII and TβRI activation results in endocytosis of the receptor complex followed by phosphorylation of substrate proteins by TβRI. In general, there are two different ways of internalization involving clathrin-coated pits (ccp, early endosomal antigen (EEA)-1+) or alternatively caveolin-coated pits (lipid rafts, caveolin-1+). It is assumed that localization to ccp leads to Smad activation, whereas routing to lipid rafts leads to degradation of receptors or activation of non-Smad signaling [18,19]. In endothelial cells (EC), Endoglin is localized to the membrane of caveolae, where it regulates the stability and interaction of eNOS and its allosteric regulator Hsp90 [20]. In addition, Endoglin was found in detergent-insoluble proteins (representing most likely lipid rafts) in placentas of pre-eclamptic women [21]. Activated Smads translocate to the nucleus in complex with the common Smad4, and in concert with positive and negative transcription factors regulate a wide spectrum of target genes [22]. In ECs, Endoglin modulates diverse functions, including migration and proliferation, by differentially regulating Smad2/3 signaling versus Smad1/5/8 signaling, which involves the interaction of Endoglin with the structural protein GIPC [13,23]. Moreover, the phosphorylation of the C-terminal part of Endoglin generates docking sites for intracellular interacting proteins, which also applies to subsets of T-cells (see below) [24,25]. However, the intracellular signal transfer modalities to regulate transcription or influence cellular functions by Endoglin (at least in EC) are not restricted to Smad-dependent mechanisms [26,27].
Endoglin expression is upregulated in activated ECs and this is facilitated by different transcription factors, including TGF-β1 regulated Smad3/Sp1/KLF6 and hypoxia regulated Hif1α [28,29]. Due to the high expression of Endoglin in activated ECs, the function and biology of Endoglin has been analyzed in detail in ECs and processes based on angiogenesis including carcinogenesis [30]. Especially, Endoglin has been exploited to target anti-cancer drugs to the tumor vasculature to treat cancer [31,32]. In contrast to EC, in tumor cells of different origins, in which Endoglin acts as a tumor suppressor, its expression is epigenetically silenced/switched off [33,34,35,36].
The area of EC research was enforced by the finding that the human disease hereditary hemorrhagic telangiectasia 1 (HHT-1) is based on genetic defects in the endoglin gene (Eng), leading to defect/non-functional or mis-targeted proteins causing haploinsufficiency in Endoglin [1,37,38,39,40]. Several Endoglin mutants have been shown to get stuck in the endoplasmic reticulum due to folding/glycosylation defects [41]. Those proteins are most likely ubiquitinated, a fact substantiated by the interaction with the E3 ubiquitin ligase tripartite motif-containing protein 21 (TRIM21) [42]. Likewise, to the multiple and sometimes life-threatening complications in HHT-1 patients, the loss of Endoglin causes embryonic lethality in homozygous Endoglin knockout mice, underpinning the important function of Endoglin during development [43]. More importantly, Endoglin knockout mouse models revealed that not only EC but also vessel-associated cells express Endoglin, and the communication of these cells with EC is the basis for defective angiogenesis [44]. In addition, the function of Endoglin is not limited to Endoglin expressing cells, since this receptor itself can directly be a part of the paracrine communication by the proteolytical release of the extracellular domain as soluble Endoglin (sEng) from the cell surface [45]. sEng is increased in pre-eclamptic woman and has been functionally linked to this disease [46]. Moreover, sEng and the membrane bound full-length Endoglin can be released by cells as a cargo of exosomes [4].
2. Endoglin in Pathological Conditions
As discussed, Endoglin is highly expressed in active/angiogenic ECs. Therefore, dysfunction of this receptor affects several organ systems relying on angiogenesis.
2.1. The Female Reproductive System
In the mouse, endoglin mRNA and protein could be localized to blood vessels and capillaries (EC) as well as interstitial fibroblasts in several tissues, [47]. Endoglin is highly expressed in the ovary, uterus and the cDNA has been cloned from a placenta library [47]. During placental development, the establishment of the fetal-maternal interaction is critical for successful pregnancy [48]. Abnormalities of placenta formation due to shallow trophoblast invasion have been linked to pre-eclampsia and fetal/intrauterine growth restriction (IUPR) [49]. Pre-eclampsia is characterized by the onset of hypertension and proteinuria in the third trimester of pregnancy and the severe forms can lead to the HELLP (Haemolysis, Elevated Liver enzyme levels, Low Platelet count) syndrome and IUPR [50,51]. The disease is based on excess placental-derived soluble VEGF receptor (sVEGFR1, sFlt1) as well as an excess of sEng released from endothelial membrane bound L-Endoglin, of which both, i.e., sFlt1 and sEng, are found elevated in the serum of pre-eclamptic women [46,52]. The liberation of sFlt1 and sEng prevents binding of their cognate ligands to EC, thereby affecting vascular tone and tissue oxygenation. In line, IUPR is often caused by hypoxia and is likely resulting from shallow trophoblast invasion in the decidua and myometrium and failure to invade the spiral arteries, a key requirement to establish an efficient utero-placental circulation [53].
In addition to EC, during pregnancy, Endoglin is permanently highly expressed on syncytiotrophoblasts and is also upregulated on extravillous trophoblasts differentiating along the invasive pathway [47,54,55]. The analysis of human villous explants revealed that artificial downregulation of Endoglin by an antibody or antisense oligonucleotides stimulates outgrows, migration and a higher Fibronectin release [56]. As mentioned above, IUPR is accompanied by placental hypoxia and increased TGF-β3 expression. Low oxygenation induces Endoglin expression (full-length and soluble form) mediated by TGF-β3 [57]. Aside ECs and trophoblasts, the expression of Endoglin was upregulated in decidua basalis of mid pregnancy in macrophages [58].
2.2. Vascular Homeostasis
The fact that HHT-1 is based on Endoglin mutations and HHT-2 is based on ALK1 mutations, both components of the TGF-β-pathway, account for ~85% of clinically diagnosed HHT patients implies that TGF-β is a critical component in this disease [59,60,61,62]. Comparative gene array analysis of human umbilical vein endothelial cells (HUVECs) from HHT-1 patients and control newborns showed several differentially expressed genes, comprising functions in extracellular matrix formation, angiogenesis, cellular adhesion and affecting genes involved in TGF-β signaling (i.e., Smad1, Smad7, and TGF-β2) [63]. Nevertheless, gene expression profiling of human nasal telangiectasial tissue and non-telangiectasial tissue revealed that not only TGF-β-related candidates are differentially expressed, but Wnt signaling also seems to be affected [62].
TGF-β 1 is a critical regulator in the endothelial system, but this applies also to hematopoiesis and mature hematopoietic cells. Therefore, the expression of Endoglin in the corresponding cells is of fundamental interest [64]. A first functional link of Endoglin to the immune system, i.e., a role of Endoglin in macrophage biology, was realized quite early. In particular, it has been noted that mononuclear cell infiltrates are observed around telangiectases consisting of lymphocytes and monocytes/macrophages [65,66]. However, immunophenotypic analysis of T-, B- and NK-lymphocytes indicated that there were no quantitative or qualitative abnormalities in HHT patients. There was no activation of T-cells and normal levels of IgGs, but the activity of components of the innate immune system was affected [67]. The phagocytotic activity and/or the oxidative burst of polymorphonuclear neutrophils (PMN) and/or monocytes were shown to be reduced in HHT patients [67]. In contrast, Guilhem and colleagues found no difference in innate immunity, but lymphopenia of T-cells (CD4, CD8) and NK cells increased, and the level of IgG and IgA was found elevated [68]. In another study, the adaptive immune system showed no quantitative change in B- and T-cells, but there was a deficit in T-cells expressing Th1-relevant cytokines (IFN-γ, IL2, TNF-α) and monocytes positive for TNF-α rendering HHT patients more susceptible to infections [69].
It is known that TGF-β1 induces macrophage differentiation [70]. Moreover, neutrophil survival, chemotaxis and activation (examined by oxidative burst and phagocytosis) are increased by TGF-β1 [71]. Therefore, it is obvious that immune cell dysfunction in the absence of Endoglin might be linked to TGF-β1, a fact substantiated by the lower TGF-β1 plasma level found in heterozygous Endoglin deficient mice compared to wild-type mice [72]. A more indirect role of Endoglin in ECs with respect to inflammation is its involvement in cell-cell contacts governing vessel architecture and the recruitment of immune cells (all lineages) from the circulation (extravasation) [44,73,74]. In turn, increased expression of Endoglin, which occurs for example in the autoimmune thyroiditis Graves’ disease and psoriatic lesions, might lead to an enhanced inflammatory cell recruitment [75,76].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21239247