The term ionone is derived from “iona” (Greek for violet) which refers to the violet scent and “ketone” due to its structure. Ionones can either be chemically synthesized or endogenously produced via asymmetric cleavage of β-carotene by β-carotene oxygenase 2 (BCO2).
- ionone,biological activity,ionone derivatives
1. Chemical Structure, Physicochemical Properties and Natural Occurrence
Ionone is a ketone compound composed of 13 carbons with a monocyclic terpenoid backbone [1]. Several isomeric ionones exist in Nature, including α-ionone, β-ionone and others. Ionones are found as secondary plant metabolism products that share mevalonic acid as a common precursor [2]. They are widely present in fruits and vegetables comprising β-carotene and are found in plant oils, for example oils from Petunia hybrida, Boronia megastigma Nees and especially Viola odorata [3][4][5][6]. It can be found in cow’s milk where is passively transferred after consumption of alfalfa pasture [7][8].
α-Ionone and β-ionone are 3-buten-2-ones substituted by 2,6,6-trimethyl-2-cyclohexen-1-yl and 2,6,6-trimethyl-1-cyclohexen-1-yl groups respectively, at position 4 (Table 1) [9][10]. The ionone stereoisomers, α-ionone and β-ionone, are pale yellow-to-yellow liquids [11] with a woody floral scent. However, the former has an extra honey olfactory aspect [12]. α-Ionone and β-ionone share similar physicochemical properties [9][10][11][13][14][15][16][17]. They are both soluble in most fixed oils, alcohol and propylene glycol but insoluble/very slightly soluble in glycerin/water, respectively [11].
| α-Ionone | β-Ionone | Reference | |
|---|---|---|---|
| Chemical structure depiction | 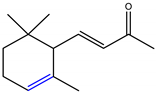 |
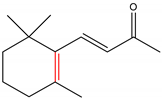 |
[9][10] |
Table 1. The chemical structure of α-ionone and β-ionone.
α-Ionone and β-ionone are both widely used as aroma ingredients in various industries, including cosmetics (e.g., shampoos, soaps) and non-cosmetic (e.g., household detergents and cleaners) products. The Flavor and Extract Manufacturer’s Association (FEMA) declares that both ionones are Generally Recognized As Safe (GRAS) when used as flavoring agents. The US FDA has also approved their use as flavoring agents [9][10].
2. Ionone Effects
2.1. β-Ionone
β-Ionone has been shown to exert a variety of pharmacological effects, including anticancer, chemopreventive, cancer-promoting, melanogenesis, anti-inflammatory and antimicrobial activity. Several mechanisms and intracellular signaling cascades have been reported explaining these effects. β-Ionone activates olfactory receptor, family 51 and subfamily E where is it member 2, (OR51E2) [18][19][20][21][22][23][24][25][26] and regulates the activity or expression of cell cycle regulatory proteins [2][27][28][29][30][31], [ pro-apoptotic and anti-apoptotic proteins [2][8][28][31][32][33][34], HMG-CoA reductase [2][29][35][36][37] and pro-inflammatory mediators [28][38]. The activation of OR51E2 causes regulation of the activity of various kinases and an increase in intracellular calcium [19][20][21][25][39]. OR51E2 activation results in anti-proliferative effect [20][21][23][24] and metastasis in prostate cancer cells [22][23][26], anti-proliferative effect and melanogenesis in melanocytes [19][25] and proliferation and metastasis in retinal pigment epithelial cells [18].
The wide spectrum of effects and various signaling cascades attributed to β-ionone might be due to the different cell repertoire [18]. In addition, β-ionone might be interacting with a wide range of receptors, not constrained to OR51E2, such as the reported interaction with AR and RXR-α [20][30]. Moreover, the hydrophobic properties of β-ionone allows it to traverse cellular membrane, activating cytosolic OR51E2 in various intracellular compartments [18][19].
2.2. α-Ionone
β-ionone acts as an agonist of OR51E2. On the other hand, α-ionone activity on OR51E2 is not as clear. Several studies reported that α-ionone antagonize OR51E2 which prevented or suppressed the effects of β-ionone, notably it has been utilized to confirm OR51E2 activation by β-ionones [19][21][22][40]. For example, Neuhaus found that α-ionone had no effect on the intracellular calcium concentration and it inhibited the β-ionone induced increase in intracellular calcium and anti-proliferative effect in prostate cancer cells [21].
In contrast, Sanz et al. showed that α-ionone is a real agonist of OR51E2. Notably, α-ionone primarily induced growth of LNCaP cells and prostate tumors in mice whereas β-ionone increased cell invasiveness. These findings are in agreement with biased agonism phenomena reported with GPCR [41]. Thus, it is proposed that each ionone induces a distinct downstream signaling cascade depending on the G protein or β-arrestin coupled [26].
In addition, α-ionone elicited distinct responses depending on its dose; moderate doses augmented LNCaP cell invasiveness while higher doses did not (instead it sustained cell growth). It is proposed that α-ionone modulates different cellular signaling cascades depending on doses. For instance, ionone might be partially or fully activating OR51E2 that triggers distinct downstream signaling cascades. It could also mean that α-ionone is antagonizing OR51E2 at higher doses. Nevertheless, the higher doses of α-ionone activating OR51E2 in transfected HEK293 cells supported the former hypothesis [26].
2.3. Ionone Derivatives
Several compounds have been synthesized and shown to exert anticancer, anti-inflammatory and antimicrobial activity.
2.3.1. Anti-Cancer
3-Hydroxy-β-ionone (Figure 1) retarded the proliferation, colony formation and cell migration of squamous cell carcinoma. In addition, it caused apoptosis and cell cycle arrest at G2/M phase in these cells. Apoptosis was indicated by an increase in cleaved caspases and Bax with a decrease in Bcl-2 [42]. It appears that 3-hydroxy-β-ionone is produced endogenously from BCO2 asymmetric cleavage of zeaxanthin, a carotenoid with a 3-hydroxy-β-ionone ring [43].
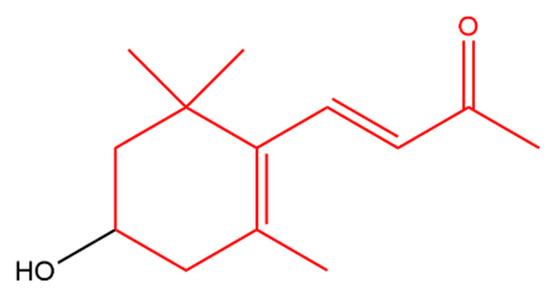
Figure 1. 3-Hydroxy-β-ionone. The β-ionone moiety is shown in red.
β-Ionone was also found to antagonize 12-O-tetradecanoylphorbol-13-acetate (TPA); a tumor-inducing compound. Another derivative, 5,6-epoxy-β-ionone (Figure 2), displayed more pronounced TPA inhibition than β-ionone in bovine lymphocytes [44].
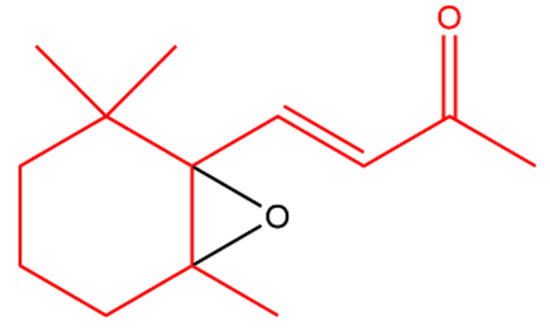
Figure 2. 5,6-Epoxy-β-ionone. The β-ionone moiety is shown in red.
Sharma and his team combined β-ionone with various aldehydes to prepare a series of chalcones. Chalcones are plant-derived ketones composed of two aromatic rings that possess anticancer and antimicrobial activity. All derivatives inhibited the proliferation of various cancer cell lines including prostate, breast, CNS, cervix and liver. Compound (a) in Figure 3 was more effective in inhibiting cell proliferation compared to other derivatives. It was noticed that compounds having electron-withdrawing group on the para position of the ring moiety were more cytotoxic than compounds with electron-donating group in the ortho position. Further studies using Chinese hamster ovary cells, showed that compound (a) caused cytotoxicity via induction of apoptosis and cell cycle arrest at G0 phase [45].
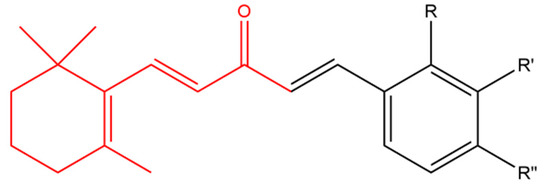
Figure 3. (Compound a). R = H, R’ = H, R’’ = NO2; [(1E,4E)-1-(4-nitrophenyl)-5-(2,6,6-trimethylcyclohex-1-en-1-yl) penta-1,4-dien-3-one]; (compound b). R = H, R’ = CF3, R’’ = H; [(1E,4E)-1-(3-(trifluoromethyl)phenyl)-5-(2,6,6-trimethylcyclohex-1-en-1-yl)penta-1,4-dien-3-one] (compound c). R = NO2, R’ = H, R” = H [(1E,4E)-1-(2-nitrophenyl)-5-(2,6,6-trimethylcyclohex-1-en-1-yl)penta-1,4-dien-3-one]; (compound d). R = Cl, R’ = H, R” = H [(1E,4E)-1-(2-chlorophenyl)-5-(2,6,6-trimethylcyclohex-1-en-1-yl)penta-1,4-dien-3-one]; (compound e). R = Br, R’ = H, R” = H; [(1E,4E)-1-(2-bromophenyl)-5-(2,6,6-trimethylcyclohex-1-en-1-yl)penta-1,4-dien-3-one]. The β-ionone moiety is shown in red.
Zhou et al. designed a variety of β-ionone-based chalcones; they exhibited various level of cytotoxic effect on a variety of prostate cancer cell lines, including LNCaP, MDA-PCa-2b, 22Rv1, C4-2B and PC-3. It is believed that compounds with an electron withdrawing group at the meta position of the ring moiety are more potent than compound with an electron donating group at the para position. Compound (b) in Figure 3 was found to induce the strongest cytotoxic effect in androgen dependent and independent prostate cancer cells even in comparison to β-ionone in the context of LNCaP and PC-3 cells. Furthermore, compound (b) was found to elicit anti-androgenic activity; as it inhibited the DHT-induced transactivation of wild type AR and various mutated ARs [46].
Zhou later synthesized a variety of β-ionone-based compounds; the compound shown in Figure 4 and compound (a) in Figure 5 were the most potent inhibitors of the proliferation of various prostate cancer cells (including LNCaP, MDA-PCa-2b, C4-2B and 22Rv1). In addition, these compounds acted as full antagonists of the wild type AR and a variety of clinically important mutated ARs in PC-3 cells [47][48]. Molecular modeling revealed that the two bulky side-chains of compound (a) in Figure 5 are critical for mutated AR antagonism. On the other hand, compound (a) elicited cytotoxic activity against PC-3 cells, which lack AR, suggesting that compound is not only targeting AR [47]. Further investigations revealed that the compounds in Figure 4 and Figure 5 inhibited NF-KB activation and interleukin-6 (IL-6) release. Compound (b) in Figure 5, a derivatives of the compound in Figure 4, also suppressed prostate tumor cell proliferation (C4-2B, 22Rv1, PC-3 and DU-145 cells), AR and NF-κB signaling. In addition, compound (b) resulted in apoptosis as it caused caspase 3 activation in LNCaP cells [49].
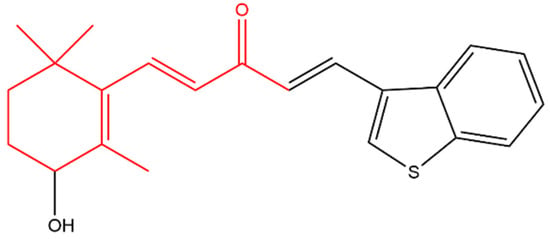
Figure 4. (1E,4E)-1-(Benzo[b]thiophen-3-yl)-5-(3-hydroxy-2,6,6-trimethylcyclohex-1-en-1-yl)penta-1,4-dien -3-one. The β-ionone moiety is shown in red.
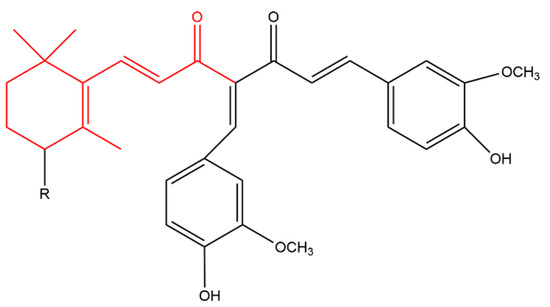
Figure 5. (Compound a). R = H; [(1E,6E)-4-((Z)-4-hydroxy-3-methoxybenzylidene)-1-(4-hydroxy-3-methoxy phenyl)-7-(2,6,6-trimethylcyclohex-1-en-1-yl)hepta-1,6-diene-3,5-dione] (compound b). R = OH; [(1E,6E)-1-(3- hydroxy-2,6,6-trimethylcyclohex-1-en-1-yl)-4-((Z)-4-hydroxy-3-methoxybenzylidene)-7-(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione]. The β-ionone moiety is shown in red.
2.3.2. Anti-Inflammatory
Balbi and co-workers synthesized heterocyclic ionone-like derivatives. However, they focused on α-ionone derivatives, as they were easier to synthesize. Almost half of the synthesized compounds inhibited superoxide anion production in neutrophils in a dose-dependent manner, in which compounds (a) and (b) (Figure 6) were found to be most active. These two compounds also inhibited the chemotactic response of neutrophils in a dose dependent manner. It was found that these derivatives inhibited neutrophil chemotaxis at lower concentrations compared to nonsteroidal anti-inflammatory drugs. It was noticed that a pyrazole ring and its substitution at position 1 seems to be very important for activity whereas an unsubstituted pyrazole (compound (a)) or an aminotriazole (compound (b)) substituent elicited the highest activity [50].
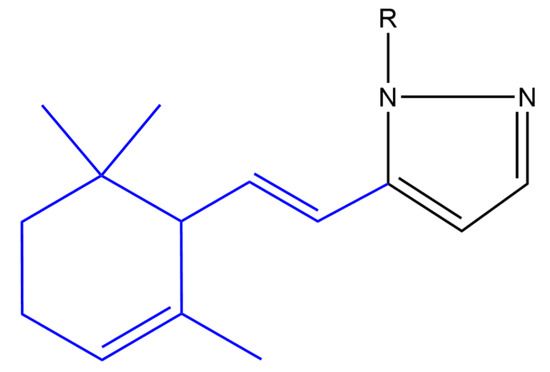
Figure 6. (Compound a). R = H; [(E)-5-(2-(2,6,6-trimethylcyclohex-2-en-1-yl)vinyl)-1H-pyrazole] (compound b). R = 4-amino-4H-1,2,4-triazole-3-thiol; [(E)-4-amino-5-(5-(2-(2,6,6-trimethylcyclohex-2-en-1-yl)vinyl)-1H-pyrazol-1 -yl)-4H-1,2,4-triazole-3-thiol] (compound c). R = phenyl [(E)-1-phenyl-5-(2-(2,6,6-trimethylcyclohex-2-en-1-yl)vinyl)-1H-pyrazole]. The α-ionone moiety is shown in blue.
In addition, a collection of β-ionone-derived curcumin analogs inhibited the secretion of pro-inflammatory cytokines including TNF-α and IL-6, following LPS stimulation in a dose dependent manner. These analogs were found to be more potent than curcumin. The most active compound (c) (Figure 3) inhibited LPS-induced septic death in mice. The study showed that having any substituent at the second position of the phenyl group resulted in normal activity against inflammatory cells while a substituent at fourth position of the phenyl group reduced the activity of the compounds. In addition, it showed that having chloro group substituents at the third and fourth position of the phenyl group displayed normal activity against inflammatory cells however replacing the chloro groups with methyl groups almost abolished this activity [51].
2.3.3. Antimicrobial Effect
A variety of heterocyclic ionone-like derivatives displayed moderate to good activity against C. albicans. Only compound (c) (Figure 6) and the compound shown in Figure 7 exhibited good activity against Pseudomonas aeruginosa, Propionibacterium acnes and E. coli. Most of the compounds showed good activity against Staphylococcus aureus. The addition of a phenyl group at the position-1 of the pyrazole ring is important for the antibacterial activity however, the substitution with 2,4-dinitrophenyl group in the same position reduced the inhibitory activity against E. coli, P. aeruginosa and S. aureus. It was concluded that lipophilicity is an important factor in increasing the activity against broader spectrum of microbial species [52].
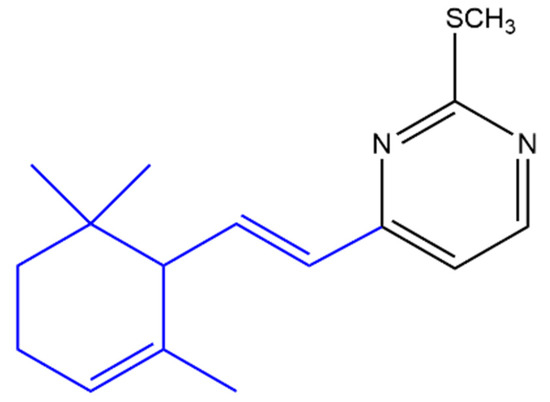
Figure 7. (E)-2-(Methylthio)-4-(2-(2,6,6-trimethylcyclohex-2-en-1-yl)vinyl)pyrimidine. The α-ionone moiety is shown in blue.
Sharma and his team combined β-ionone with various aldehydes to prepare a series of chalcones. The active compounds exhibited promising antimicrobial activity against a variety of Gram negative bacteria (E. coli, P. aeruginosa, Salmonella typhimurium), Gram positive bacteria (S. aureus, Bacillus subtilis) and fungal strains (A. niger, Saccharomyces cerevisiae, C. albicans). In addition, further testing showed that some of the active compounds were also active against methicillin resistant S. aureus. Compound (d) in Figure 3 displayed the highest anti-bacterial activity against both Gram negative and positive bacteria while compound (e) showed the uppermost antifungal activity against species including A. niger and S. cerevisiae. It was noticed that compounds with electron withdrawing substituents on the aromatic ring mostly displayed better antibacterial activity in comparison to compounds with electron releasing groups [53].
β-Ionone was also used as a starting material to produce a variety of synthetic halolactones and microbiologically obtained hydroxylactones. All of the derivatives inhibited the growth of B. subtilis, S. aureus and E.coli, however, the chlorolactone (compound (a) in Figure 8) and hydroxylactone (compound (b)) completely inhibited the growth of B. subtilis and S. aureus, respectively. In addition, all of the compounds increased the growth of A. niger and Fusarium linii except the bromolactone (compound (c) in Figure 8) which completely inhibited the growth of A. niger [54].
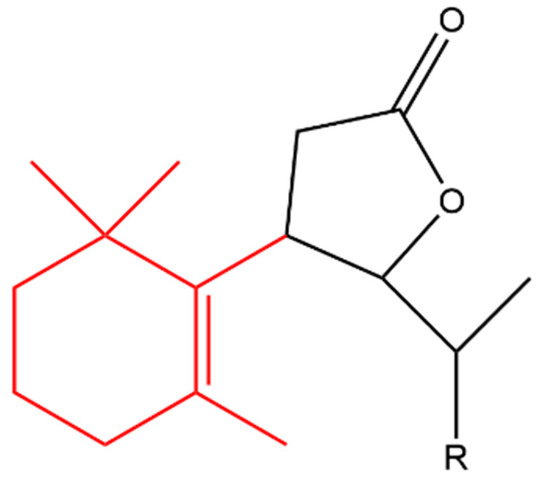
Figure 8. (Compound a). R = Cl; [10-(11-chloroethyl)-7-(1,1,5-trimethylcyclohex-5-en-6-yl)dihydrofuran-2(3H)-one]; (compound b). R = OH [10-(11-hydroxyethyl)-7-(1,1,5-trimethylcyclohex-5-en-6-yl)dihydrofuran-2(3H)-one]; (compound c). R = Br [10-(11-bromoethyl)-7-(1,1,5-trimethylcyclohex-5-en-6-yl)dihydrofuran-2(3H)-one]. The β-ionone moiety is shown in red.
Suryawanshi and his team synthesized several terpenyl pyrimidines substituted with 4-N in which the starting material (ketene dithioacetal) was made available from β-ionone. The 4-thiomethoxy substituted pyrimidine (Figure 9) caused 66% in vivo inhibition of Leishmania donovani in hamsters. It was noticed that the 4-thiomethoxy group is very important for the anti-leishmanial activity as its replacement with various primary and secondary amines resulted in activity loss [55].
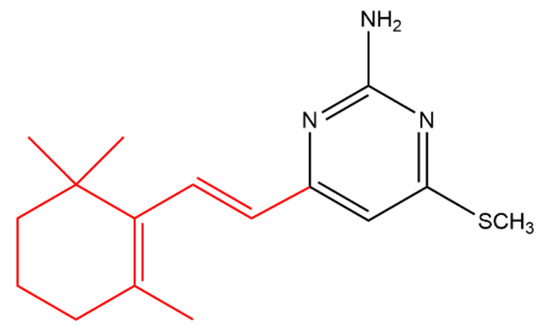
Figure 9. (E)-4-(methylthio)-6-(2-(2,6,6-trimethylcyclohex-1-en-1-yl)vinyl)pyrimidin-2-amine. The β-ionone moiety is shown in red.
This entry is adapted from the peer-reviewed paper 10.3390/molecules25245822
References
- Pinto, F.C.M.; De-Carvalho, R.R.; De-Oliveira, A.; Delgado, I.F.; Paumgartten, F.J.R. Study on the developmental toxicity of beta-ionone in the rat. Regul Toxicol Pharmacol 2018, 97, 110-119, doi:10.1016/j.yrtph.2018.06.009.
- Mo, H.; Elson, C.E. Apoptosis and cell-cycle arrest in human and murine tumor cells are initiated by isoprenoids. J Nutr 1999, 129, 804-813, doi:10.1093/jn/129.4.804.
- Simpson, K.L.; Chichester, C.O. Metabolism and nutritional significance of carotenoids. Annu Rev Nutr 1981, 1, 351-374, doi:10.1146/annurev.nu.01.070181.002031.
- Cooper, C.M.; Davies, N.W.; Menary, R.C. C-27 apocarotenoids in the flowers of Boronia megastigma (Nees). J Agric Food Chem 2003, 51, 2384-2389, doi:10.1021/jf026007c.
- Simkin, A.J.; Underwood, B.A.; Auldridge, M.; Loucas, H.M.; Shibuya, K.; Schmelz, E.; Clark, D.G.; Klee, H.J. Circadian regulation of the PhCCD1 carotenoid cleavage dioxygenase controls emission of beta-ionone, a fragrance volatile of petunia flowers. Plant Physiol 2004, 136, 3504-3514, doi:10.1104/pp.104.049718.
- Ansari, M.; Emami, S. beta-Ionone and its analogs as promising anticancer agents. Eur J Med Chem 2016, 123, 141-154, doi:10.1016/j.ejmech.2016.07.037.
- Elson, C.E.; Yu, S.G. The chemoprevention of cancer by mevalonate-derived constituents of fruits and vegetables. J Nutr 1994, 124, 607-614, doi:10.1093/jn/124.5.607.
- Liu, Q.; Dong, H.W.; Sun, W.G.; Liu, M.; Ibla, J.C.; Liu, L.X.; Parry, J.W.; Han, X.H.; Li, M.S.; Liu, J.R. Apoptosis initiation of beta-ionone in SGC-7901 gastric carcinoma cancer cells via a PI3K-AKT pathway. Arch Toxicol 2013, 87, 481-490, doi:10.1007/s00204-012-0962-8.
- Lalko, J.; Lapczynski, A.; McGinty, D.; Bhatia, S.; Letizia, C.S.; Api, A.M. Fragrance material review on beta-ionone. Food Chem Toxicol 2007, 45 Suppl 1, S241-247, doi:10.1016/j.fct.2007.09.052.
- Lalko, J.; Lapczynski, A.; Politano, V.T.; McGinty, D.; Bhatia, S.; Letizia, C.S.; Api, A.M. Fragrance material review on alpha-ionone. Food Chem Toxicol 2007, 45 Suppl 1, S235-240, doi:10.1016/j.fct.2007.09.046.
- O'Neil, M.J. The Merck index : an encyclopedia of chemicals, drugs, and biologicals. In Encyclopedia of chemicals, drugs, and biologicals, 15 ed.; Cambridge, UK : Royal Society of Chemistry: England, 2013; p 940
- Brenna E, F.C., Serra S, Kraft P Optically active ionones and derivatives: preparation and olfactory properties. Eur J Org Chem 2002, 967-978.
- Terpenoids., S.C. Kirk-Othmer Encyclopedia of Chemical Technology. Fourth ed.; Kirk-Othmer, Ed. John Wiley & Sons. : New York, USA, 2007.
- Showing Compound alpha-Ionone (FDB014484). Availabe online: https://foodb.ca/compounds/FDB014484 (accessed on 10 - November).
- Lide, D.R., G.W.A. Milne Handbook of Data on Organic Compounds, 3 ed.; CRC Press: 1994; Vol. I.
- Etzweiler, F.; Senn, E.; Schmidt, H.W.H. Method for Measuring Aqueous Solubilities of Organic Compounds. Analytical Chemistry 1995, 67, 655-658, doi:10.1021/ac00099a025.
- Fichan, I.; Larroche, C.; Gros, J.B. Water Solubility, Vapor Pressure, and Activity Coefficients of Terpenes and Terpenoids. Journal of Chemical & Engineering Data 1999, 44, 56-62, doi:10.1021/je980070+.
- Jovancevic, N.; Khalfaoui, S.; Weinrich, M.; Weidinger, D.; Simon, A.; Kalbe, B.; Kernt, M.; Kampik, A.; Gisselmann, G.; Gelis, L., et al. Odorant Receptor 51E2 Agonist β-ionone Regulates RPE Cell Migration and Proliferation. Frontiers in physiology 2017, 8, 888, doi:10.3389/fphys.2017.00888.
- Gelis, L.; Jovancevic, N.; Veitinger, S.; Mandal, B.; Arndt, H.D.; Neuhaus, E.M.; Hatt, H. Functional Characterization of the Odorant Receptor 51E2 in Human Melanocytes. The Journal of biological chemistry 2016, 291, 17772-17786, doi:10.1074/jbc.M116.734517.
- Xie, H.; Liu, T.; Chen, J.; Yang, Z.; Xu, S.; Fan, Y.; Zeng, J.; Chen, Y.; Ma, Z.; Gao, Y., et al. Activation of PSGR with β-ionone suppresses prostate cancer progression by blocking androgen receptor nuclear translocation. Cancer letters 2019, 453, 193-205, doi:10.1016/j.canlet.2019.03.044.
- Neuhaus, E.M.; Zhang, W.; Gelis, L.; Deng, Y.; Noldus, J.; Hatt, H. Activation of an olfactory receptor inhibits proliferation of prostate cancer cells. The Journal of biological chemistry 2009, 284, 16218-16225, doi:10.1074/jbc.M109.012096.
- Sanz, G.; Leray, I.; Dewaele, A.; Sobilo, J.; Lerondel, S.; Bouet, S.; Grébert, D.; Monnerie, R.; Pajot-Augy, E.; Mir, L.M. Promotion of cancer cell invasiveness and metastasis emergence caused by olfactory receptor stimulation. PLoS One 2014, 9, e85110, doi:10.1371/journal.pone.0085110.
- Cao, W.; Li, F.; Yao, J.; Yu, J. Prostate specific G protein coupled receptor is associated with prostate cancer prognosis and affects cancer cell proliferation and invasion. BMC cancer 2015, 15, 915, doi:10.1186/s12885-015-1921-6.
- Wiese, H.; Gelis, L.; Wiese, S.; Reichenbach, C.; Jovancevic, N.; Osterloh, M.; Meyer, H.E.; Neuhaus, E.M.; Hatt, H.H.; Radziwill, G., et al. Quantitative phosphoproteomics reveals the protein tyrosine kinase Pyk2 as a central effector of olfactory receptor signaling in prostate cancer cells. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2015, 1854, 632-640, doi:https://doi.org/10.1016/j.bbapap.2014.09.002.
- Gelis, L.; Jovancevic, N.; Bechara, F.G.; Neuhaus, E.M.; Hatt, H. Functional expression of olfactory receptors in human primary melanoma and melanoma metastasis. Experimental dermatology 2017, 26, 569-576, doi:10.1111/exd.13316.
- Sanz, G.; Leray, I.; Grébert, D.; Antoine, S.; Acquistapace, A.; Muscat, A.; Boukadiri, A.; Mir, L.M. Structurally related odorant ligands of the olfactory receptor OR51E2 differentially promote metastasis emergence and tumor growth. Oncotarget 2017, 8, 4330-4341, doi:10.18632/oncotarget.13836.
- Duncan, R.E.; Lau, D.; El-Sohemy, A.; Archer, M.C. Geraniol and beta-ionone inhibit proliferation, cell cycle progression, and cyclin-dependent kinase 2 activity in MCF-7 breast cancer cells independent of effects on HMG-CoA reductase activity. Biochemical pharmacology 2004, 68, 1739-1747, doi:10.1016/j.bcp.2004.06.022.
- Dong, H.W.; Wang, K.; Chang, X.X.; Jin, F.F.; Wang, Q.; Jiang, X.F.; Liu, J.R.; Wu, Y.H.; Yang, C. Beta-ionone-inhibited proliferation of breast cancer cells by inhibited COX-2 activity. Arch Toxicol 2019, 93, 2993-3003, doi:10.1007/s00204-019-02550-2.
- Jones, S.; Fernandes, N.V.; Yeganehjoo, H.; Katuru, R.; Qu, H.; Yu, Z.; Mo, H. β-ionone induces cell cycle arrest and apoptosis in human prostate tumor cells. Nutrition and cancer 2013, 65, 600-610, doi:10.1080/01635581.2013.776091.
- Janakiram, N.B.; Cooma, I.; Mohammed, A.; Steele, V.E.; Rao, C.V. Beta-ionone inhibits colonic aberrant crypt foci formation in rats, suppresses cell growth, and induces retinoid X receptor-alpha in human colon cancer cells. Molecular cancer therapeutics 2008, 7, 181-190, doi:10.1158/1535-7163.mct-07-0529.
- Liu, J.-R.; Sun, X.-R.; Dong, H.-W.; Sun, C.-H.; Sun, W.-G.; Chen, B.-Q.; Song, Y.-Q.; Yang, B.-F. β-Ionone suppresses mammary carcinogenesis, proliferative activity and induces apoptosis in the mammary gland of the Sprague-Dawley rat. International Journal of Cancer 2008, 122, 2689-2698, doi:https://doi.org/10.1002/ijc.23453.
- Liu, J.R.; Chen, B.Q.; Yang, B.F.; Dong, H.W.; Sun, C.H.; Wang, Q.; Song, G.; Song, Y.Q. Apoptosis of human gastric adenocarcinoma cells induced by beta-ionone. World journal of gastroenterology 2004, 10, 348-351, doi:10.3748/wjg.v10.i3.348.
- Sun, X.; Liu, J.; Sun, W.; Tang, X. [Beta-ionone induced apoptosis in SGC-7901 cells]. Wei sheng yan jiu = Journal of hygiene research 2007, 36, 667-670.
- Dong, H.W.; Zhang, S.; Sun, W.G.; Liu, Q.; Ibla, J.C.; Soriano, S.G.; Han, X.H.; Liu, L.X.; Li, M.S.; Liu, J.R. β-Ionone arrests cell cycle of gastric carcinoma cancer cells by a MAPK pathway. Arch Toxicol 2013, 87, 1797-1808, doi:10.1007/s00204-013-1041-5.
- Yu, S.G.; Abuirmeileh, N.M.; Qureshi, A.A.; Elson, C.E. Dietary .beta.-ionone suppresses hepatic 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Journal of Agricultural and Food Chemistry 1994, 42, 1493-1496, doi:10.1021/jf00043a019.
- Case, G.L.; He, L.; Mo, H.; Elson, C.E. Induction of geranyl pyrophosphate pyrophosphatase activity by cholesterol-suppressive isoprenoids. Lipids 1995, 30, 357-359, doi:10.1007/bf02536045.
- de Moura Espíndola, R.; Mazzantini, R.P.; Ong, T.P.; de Conti, A.; Heidor, R.; Moreno, F.S. Geranylgeraniol and beta-ionone inhibit hepatic preneoplastic lesions, cell proliferation, total plasma cholesterol and DNA damage during the initial phases of hepatocarcinogenesis, but only the former inhibits NF-kappaB activation. Carcinogenesis 2005, 26, 1091-1099, doi:10.1093/carcin/bgi047.
- Kang, C.H.; Jayasooriya, R.G.; Choi, Y.H.; Moon, S.K.; Kim, W.J.; Kim, G.Y. β-Ionone attenuates LPS-induced pro-inflammatory mediators such as NO, PGE2 and TNF-α in BV2 microglial cells via suppression of the NF-κB and MAPK pathway. Toxicology in vitro : an international journal published in association with BIBRA 2013, 27, 782-787, doi:10.1016/j.tiv.2012.12.012.
- Spehr, J.; Gelis, L.; Osterloh, M.; Oberland, S.; Hatt, H.; Spehr, M.; Neuhaus, E.M. G protein-coupled receptor signaling via Src kinase induces endogenous human transient receptor potential vanilloid type 6 (TRPV6) channel activation. The Journal of biological chemistry 2011, 286, 13184-13192, doi:10.1074/jbc.M110.183525.
- Maßberg, D.; Hatt, H. Human Olfactory Receptors: Novel Cellular Functions Outside of the Nose. Physiological reviews 2018, 98, 1739-1763, doi:10.1152/physrev.00013.2017.
- Luttrell, L.M.; Maudsley, S.; Bohn, L.M. Fulfilling the Promise of "Biased" G Protein-Coupled Receptor Agonism. Molecular pharmacology 2015, 88, 579-588, doi:10.1124/mol.115.099630.
- Luetragoon, T.; Pankla Sranujit, R.; Noysang, C.; Thongsri, Y.; Potup, P.; Suphrom, N.; Nuengchamnong, N.; Usuwanthim, K. Anti-Cancer Effect of 3-Hydroxy-β-Ionone Identified from Moringa oleifera Lam. Leaf on Human Squamous Cell Carcinoma 15 Cell Line. Molecules (Basel, Switzerland) 2020, 25, doi:10.3390/molecules25163563.
- Lobo, G.P.; Isken, A.; Hoff, S.; Babino, D.; von Lintig, J. BCDO2 acts as a carotenoid scavenger and gatekeeper for the mitochondrial apoptotic pathway. Development (Cambridge, England) 2012, 139, 2966-2977, doi:10.1242/dev.079632.
- Wertz, P.W.; Kensler, T.W.; Mueller, G.C. Inhibition of phorbol ester action in lymphocytes by 5,6-epoxy-β-ionone. Biochemical and Biophysical Research Communications 1978, 83, 138-143, doi:https://doi.org/10.1016/0006-291X(78)90408-4.
- Sharma, V.; Chaudhary, A.; Arora, S.; Saxena, A.K.; Ishar, M.P.S. β-Ionone derived chalcones as potent antiproliferative agents. European Journal of Medicinal Chemistry 2013, 69, 310-315, doi:https://doi.org/10.1016/j.ejmech.2013.08.017.
- Zhou, J.; Geng, G.; Batist, G.; Wu, J.H. Syntheses and potential anti-prostate cancer activities of ionone-based chalcones. Bioorganic & medicinal chemistry letters 2009, 19, 1183-1186, doi:10.1016/j.bmcl.2008.12.089.
- Zhou, J.; Geng, G.; Shi, Q.; Sauriol, F.; Wu, J.H. Design and synthesis of androgen receptor antagonists with bulky side chains for overcoming antiandrogen resistance. Journal of medicinal chemistry 2009, 52, 5546-5550, doi:10.1021/jm801218k.
- Zhou, J.; Geng, G.; Wu, J.H. Synthesis and in vitro characterization of ionone-based chalcones as novel antiandrogens effective against multiple clinically relevant androgen receptor mutants. Investigational new drugs 2010, 28, 291-298, doi:10.1007/s10637-009-9251-7.
- Liu, W.; Zhou, J.; Geng, G.; Lin, R.; Wu, J.H. Synthesis and in vitro characterization of ionone-based compounds as dual inhibitors of the androgen receptor and NF-κB. Investigational new drugs 2014, 32, 227-234, doi:10.1007/s10637-013-0040-y.
- Balbi, A.; Anzaldi, M.; Mazzei, M.; Miele, M.; Bertolotto, M.; Ottonello, L.; Dallegri, F. Synthesis and biological evaluation of novel heterocyclic ionone-like derivatives as anti-inflammatory agents. Bioorganic & Medicinal Chemistry 2006, 14, 5152-5160, doi:https://doi.org/10.1016/j.bmc.2006.04.007.
- Hu, W.; Cai, M.; Qi, D.; Ying, X.; Huang, C.; Xing, C. β-Ionone-Derived Curcumin Analogs as Potent Anti-Inflammatory Agents. Pharmaceutical Chemistry Journal 2018, 51, 902-906, doi:10.1007/s11094-018-1713-9.
- Anzaldi, M.; Sottofattori, E.; Rizzetto, R.; Granello di Casaleto, B.; Balbi, A. Synthesis and antimicrobial activity of heterocyclic ionone-like derivatives. European Journal of Medicinal Chemistry 1999, 34, 837-842, doi:https://doi.org/10.1016/S0223-5234(99)00209-3.
- Sharma, V.; Singh, G.; Kaur, H.; Saxena, A.K.; Ishar, M.P. Synthesis of β-ionone derived chalcones as potent antimicrobial agents. Bioorganic & medicinal chemistry letters 2012, 22, 6343-6346, doi:10.1016/j.bmcl.2012.08.084.
- Grabarczyk, M.; Wińska, K.; Mączka, W.; Żarowska, B.; Maciejewska, G.; Dancewicz, K.; Gabryś, B.; Anioł, M. Synthesis, biotransformation and biological activity of halolactones obtained from β-ionone. Tetrahedron 2016, 72, 637-644, doi:https://doi.org/10.1016/j.tet.2015.12.005.
- Suryawanshi, S.N.; Bhat, B.A.; Pandey, S.; Chandra, N.; Gupta, S. Chemotherapy of leishmaniasis. Part VII: synthesis and bioevaluation of substituted terpenyl pyrimidines. Eur J Med Chem 2007, 42, 1211-1217, doi:10.1016/j.ejmech.2006.10.002.