Anti-SARS-CoV-2 vaccines have played a pivotal role in reducing the risk of developing severe illness from COVID-19, thus helping end the COVID-19 global public health emergency. Intriguingly, as SARS-CoV-2 variants emerged, individuals who were fully vaccinated did get infected in high numbers, and viral loads in vaccinated individuals were as high as those in the unvaccinated. However, vaccines undoubtedly offered protection against severe illness even without conferring immunity in the classical sense. The lessons learned from anti-SARS-CoV-2 vaccines are a call to the medical community to revisit and redefine the concept of vaccine effectiveness. This endeavor is likely to increase increase vaccine confidence, and thus bolster global health education efforts and preventive care.
- SARS-CoV-2
- immunity
- cytokines
- infectious diseases
1. Introduction
2. SARS-CoV-2—Killer Virus or Just a Trigger for Kitchen Sink Inflammation?
2.1. Epithelial System
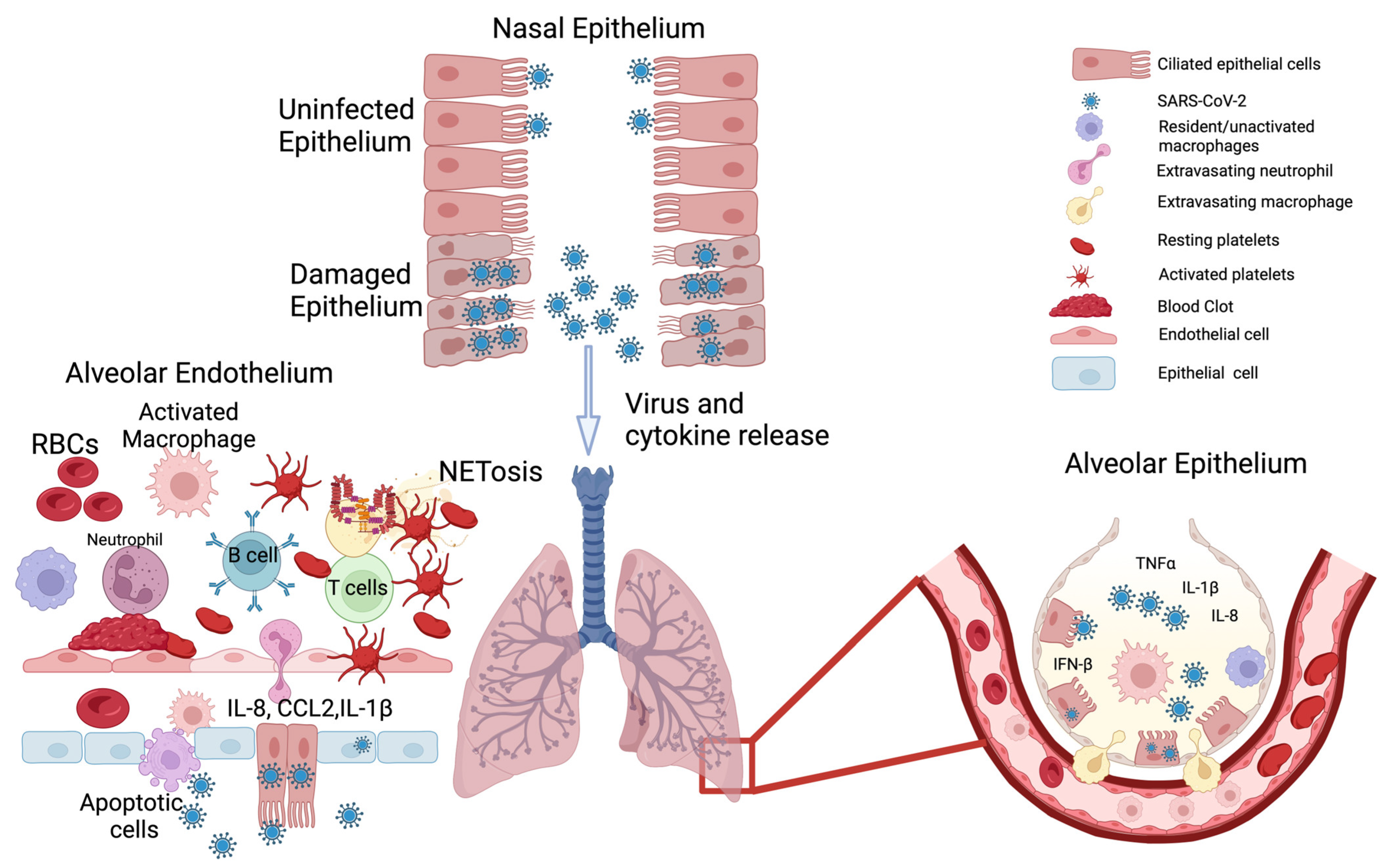
2.2. Endothelial System
2.3. The Mismatch between Viral Load and Symptom Severity

This entry is adapted from the peer-reviewed paper 10.3390/microorganisms11081963
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154.
- World Health Organization (WHO). Statement on the Fifteenth Meeting of the Ihr (2005) Emergency Committee on the COVID-19 Pandemic; World Health Organization (WHO): Geneva, Switzerland, 2005; Available online: https://www.who.int/news/item/05-05-2023-statement-on-the-fifteenth-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-coronavirus-disease-(COVID-19)-pandemic (accessed on 11 May 2023).
- Wise, J. COVID-19: Who Declares End of Global Health Emergency. Bmj 2023, 381, 1041.
- Lenharo, M. Who Declares End to COVID-19’s Emergency Phase. Nature 2023.
- Ranney, M.L.; Valerie Griffeth, M.P.H.; Jha, A.K. Critical Supply Shortages—The Need for Ventilators and Personal Protective Equipment during the COVID-19 Pandemic. N. Engl. J. Med. 2020, 382, e41.
- Dey, A.K.; Bhan, N.; Rao, N.; Ghule, M.; Chatterji, S.; Raj, A. Factors Affecting Delayed and Non-Receipt of Healthcare During the COVID-19 Pandemic for Women in Rural Maharashtra, India: Evidence from a Cross-Sectional Study. EClinicalMedicine 2022, 53, 101741.
- Rogers, E.H.; Porter, J.N.; Ren, J.J.; Battista, J.J.; Grewal, N.P.; Valentin, N.I. Experiences from the Epicenter: An Observational Report on Converting a Post Anesthesia Care Unit to a COVID-19 Intensive Care Unit. Int. J. Surg. Glob. Health 2020, 3, e20.
- Davis, B.; Bankhead-Kendall, B.K.; Dumas, R.P. A Review of COVID-19’s Impact on Modern Medical Systems from a Health Organization Management Perspective. Health Technol. 2022, 12, 815–824.
- Parotto, E.; Lamberti-Castronuovo, A.; Censi, V.; Valente, M.; Atzori, A.; Ragazzoni, L. Exploring Italian Healthcare Facilities Response to COVID-19 Pandemic: Lessons Learned from the Italian Response to COVID-19 Initiative. Front. Public Health 2022, 10, 1016649.
- The Lancet Public, Health. COVID-19 in Spain: A Predictable Storm? Lancet Public Health 2020, 5, e568.
- Sharma, A.; Gupta, P.; Jha, R. COVID-19: Impact on Health Supply Chain and Lessons to Be Learnt. J. Health Manag. 2020, 22, 248–261.
- Usher, A.D. Medical Oxygen Crisis: A Belated COVID-19 Response. Lancet 2021, 397, 868–869.
- Feinmann, J. How COVID-19 Revealed the Scandal of Medical Oxygen Supplies Worldwide. Bmj 2021, 373, n1166.
- Tenforde, M.W.; Self, W.H.; Adams, K.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Association between Mrna Vaccination and COVID-19 Hospitalization and Disease Severity. JAMA 2021, 326, 2043–2054.
- Moghadas, S.M.; Vilches, T.N.; Zhang, K.; Wells, C.R.; Shoukat, A.; Singer, B.H.; Meyers, L.A.; Neuzil, K.M.; Langley, J.M.; Fitzpatrick, M.C.; et al. The Impact of Vaccination on COVID-19 Outbreaks in the United States. medRxiv 2021.
- Watson, O.J.; Barnsley, G.; Toor, J.; Hogan, A.B.; Winskill, P.; Ghani, A.C. Ghani. Global Impact of the First Year of COVID-19 Vaccination: A Mathematical Modelling Study. Lancet Infect. Dis. 2022, 22, 1293–1302.
- Torreele, E.; Kazatchkine, M.; Liu, J.; Dybul, M.; Cárdenas, M.; Singh, S.; Quigley, H.L.; McNab, C.; Sirleaf, E.J.; Mazzucato, M.; et al. Stopping Epidemics When and Where They Occur. Lancet 2023, 401, 324–328.
- Samudrala, P.K.; Kumar, P.; Choudhary, K.; Thakur, N.; Wadekar, G.S.; Dayaramani, R.; Agrawal, M.; Alexander, A. Virology, Pathogenesis, Diagnosis and in-Line Treatment of COVID-19. Eur. J. Pharmacol. 2020, 883, 173375.
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, Epidemiology, Pathogenesis, and Control of COVID-19. Viruses 2020, 12, 372.
- Ciotti, M.; Ciccozzi, M.; Terrinoni, A.; Jiang, W.C.; Wang, C.B.; Bernardini, S. The COVID-19 Pandemic. Crit. Rev. Clin. Lab Sci. 2020, 57, 365–388.
- Huang, H.; Cai, S.; Li, Y.; Li, Y.; Fan, Y.; Li, L.; Lei, C.; Tang, X.; Hu, F.; Li, F.; et al. Prognostic Factors for COVID-19 Pneumonia Progression to Severe Symptoms Based on Earlier Clinical Features: A Retrospective Analysis. Front. Med. 2020, 7, 557453.
- Morrow, A.J.; Sykes, R.; McIntosh, A.; Kamdar, A.; Bagot, C.; Bayes, H.K.; Blyth, K.G.; Briscoe, M.; Bulluck, H.; Carrick, D.; et al. A Multisystem, Cardio-Renal Investigation of Post-COVID-19 Illness. Nat. Med. 2022, 28, 1303–1313.
- Gupta, A.; Madhavan, M.V.; Sehgal, K.; Nair, N.; Mahajan, S.; Sehrawat, T.S.; Bikdeli, B.; Ahluwalia, N.; Ausiello, J.C.; Wan, E.Y.; et al. Extrapulmonary Manifestations of COVID-19. Nat. Med. 2020, 26, 1017–1032.
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. Histochem. J. 2020, 51, 613–628.
- Que, Y.; Hu, C.; Wan, K.; Hu, P.; Wang, R.; Luo, J.; Li, T.; Ping, R.; Hu, Q.; Sun, Y.; et al. Cytokine Release Syndrome in COVID-19: A Major Mechanism of Morbidity and Mortality. Int. Rev. Immunol. 2022, 41, 217–230.
- Montazersaheb, S.; Khatibi, S.M.H.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Sorbeni, F.G.; Farahzadi, R.; Ghasemnejad, T. Ghasemian Sorbeni, R. Farahzadi, and T. Ghasemnejad. COVID-19 Infection: An Overview on Cytokine Storm and Related Interventions. Virol. J. 2022, 19, 92.
- Mattoo SU, S.; Kim, S.J.; Ahn, D.G.; Myoung, J. Escape and over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin. Viruses 2022, 14, 530.
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An Introduction to Immunology and Immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49.
- Murgolo, N.; Therien, A.G.; Howell, B.; Klein, D.; Koeplinger, K.; Lieberman, L.A.; Adam, G.C.; Flynn, J.; McKenna, P.; Swaminathan, G.; et al. SARS-CoV-2 tropism, entry, replication, and propagation: Considerations for drug discovery and development. PLoS Pathog. 2021, 17, e1009225.
- Gengler, I.; Wang, J.C.; Speth, M.M.; Sedaghat, A.R. Sinonasal pathophysiology of SARS-CoV-2 and COVID-19: A systematic review of the current evidence. Laryngoscope Investig. Otolaryngol. 2020, 5, 354–359.
- Wruck, W.; Adjaye, J. SARS-CoV-2 receptor ACE2 is co-expressed with genes related to transmembrane serine proteases, viral entry, immunity and cellular stress. Sci. Rep. 2020, 10, 21415.
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687.
- Collin, J.; Queen, R.; Zerti, D.; Dorgau, B.; Georgiou, M.; Djidrovski, I.; Hussain, R.; Coxhead, J.M.; Joseph, A.; Rooney, P.; et al. Co-expression of SARS-CoV-2 entry genes in the superficial adult human conjunctival, limbal and corneal epithelium suggests an additional route of entry via the ocular surface. Ocul. Surf. 2021, 19, 190–200.
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170.
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156.
- Gamage, A.M.; Tan, K.S.; Chan, W.O.Y.; Liu, J.; Tan, C.W.; Ong, Y.K.; Thong, M.; Andiappan, A.K.; Anderson, D.E.; Wang, Y.; et al. Infection of human Nasal Epithelial Cells with SARS-CoV-2 and a 382-nt deletion isolate lacking ORF8 reveals similar viral kinetics and host transcriptional profiles. PLoS Pathog. 2020, 16, e1009130.
- Zhu, N.; Wang, W.; Liu, Z.; Liang, C.; Wang, W.; Ye, F.; Huang, B.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat. Commun. 2020, 11, 3910.
- Ozturk, E.O.; Aslan, M.; Bayindir, T. The effect of COVID-19 on nasal mucociliary clearance. Acta Otolaryngol. 2022, 142, 329–332.
- Thomas, B.; Rutman, A.; Hirst, R.A.; Haldar, P.; Wardlaw, A.J.; Bankart, J.; Brightling, C.E.; O’Callaghan, C. Ciliary dysfunction and ultrastructural abnormalities are features of severe asthma. J. Allergy Clin. Immunol. 2010, 126, 722–729.e2.
- Leung, H.M.; Birket, S.E.; Hyun, C.; Ford, T.N.; Cui, D.; Solomon, G.M.; Shei, R.J.; Adewale, A.T.; Lenzie, A.R.; Fernandez-Petty, C.M.; et al. Intranasal micro-optical coherence tomography imaging for cystic fibrosis studies. Sci. Transl. Med. 2019, 11, eaav3505.
- Yaghi, A.; Zaman, A.; Cox, G.; Dolovich, M.B. Ciliary beating is depressed in nasal cilia from chronic obstructive pulmonary disease subjects. Respir. Med. 2012, 106, 1139–1147.
- Fiege, J.K.; Thiede, J.M.; Nanda, H.A.; Matchett, W.E.; Moore, P.J.; Montanari, N.R.; Thielen, B.K.; Daniel, J.; Stanley, E.; Hunter, R.C.; et al. Single cell resolution of SARS-CoV-2 tropism, antiviral responses, and susceptibility to therapies in primary human airway epithelium. PLoS Pathog. 2021, 17, e1009292.
- Galani, I.-E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40.
- Kim, Y.-M.; Shin, E.-C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760.
- Ziegler, C.G.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733.e4722.
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284.
- Bosmuller, H.; Matter, M.; Fend, F.; Tzankov, A. The pulmonary pathology of COVID-19. Virchows Arch. 2021, 478, 137–150.
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192.
- Huang, J.; Hume, A.J.; Abo, K.M.; Werder, R.B.; Villacorta-Martin, C.; Alysandratos, K.D.; Beermann, M.L.; Simone-Roach, C.; Lindstrom-Vautrin, J.; Olejnik, J.; et al. SARS-CoV-2 Infection of Pluripotent Stem Cell-Derived Human Lung Alveolar Type 2 Cells Elicits a Rapid Epithelial-Intrinsic Inflammatory Response. Cell Stem Cell 2020, 27, 962–973.e967.
- Rockx, B.; Kuiken, T.; Herfst, S.; Bestebroer, T.; Lamers, M.M.; Oude Munnink, B.B.; de Meulder, D.; van Amerongen, G.; van den Brand, J.; Okba, N.M.A.; et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020, 368, 1012–1015.
- Queiroz, M.A.F.; Neves, P.; Lima, S.S.; Lopes, J.D.C.; Torres, M.; Vallinoto, I.; Bichara, C.D.A.; Dos Santos, E.F.; de Brito, M.; da Silva, A.L.S.; et al. Cytokine Profiles Associated with Acute COVID-19 and Long COVID-19 Syndrome. Front. Cell Infect. Microbiol. 2022, 12, 922422.
- Wilson, J.G.; Simpson, L.J.; Ferreira, A.-M.; Rustagi, A.; Roque, J.; Asuni, A.; Ranganath, T.; Grant, P.M.; Subramanian, A.; Rosenberg-Hasson, Y.; et al. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight 2020, 5, 289.
- Huang, I.; Pranata, R.; Lim, M.A.; Oehadian, A.; Alisjahbana, B. C-reactive protein, procalcitonin, D-dimer, and ferritin in severe coronavirus disease-2019: A meta-analysis. Ther. Adv. Respir. Dis. 2020, 14, 1753466620937175.
- Bello, S.; Lasierra, A.B.; López-Vergara, L.; de Diego, C.; Torralba, L.; de Gopegui, P.R.; Lahoz, R.; Abadía, C.; Godino, J.; Cebollada, A.; et al. IL-6 and cfDNA monitoring throughout COVID-19 hospitalization are accurate markers of its outcomes. Respir. Res. 2023, 24, 125.
- Frisoni, P.; Neri, M.; D’Errico, S.; Alfieri, L.; Bonuccelli, D.; Cingolani, M.; Di Paolo, M.; Gaudio, R.M.; Lestani, M.; Marti, M.; et al. Cytokine storm and histopathological findings in 60 cases of COVID-19-related death: From viral load research to immunohistochemical quantification of major players IL-1β, IL-6, IL-15 and TNF-α. Forensic. Sci. Med. Pathol. 2022, 18, 4–19.
- Meyer, N.J.; Gattinoni, L.; Calfee, C.S. Acute respiratory distress syndrome. Lancet 2021, 398, 622–637.
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473.
- Grant, R.A.; Morales-Nebreda, L.; Markov, N.S.; Swaminathan, S.; Querrey, M.; Guzman, E.R.; Abbott, D.A.; Donnelly, H.K.; Donayre, A.; Goldberg, I.A.; et al. Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 2021, 590, 635–641.
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979.
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The spatial landscape of lung pathology during COVID-19 progression. Nature 2021, 593, 564–569.
- Galani, I.E.; Andreakos, E. Neutrophils in viral infections: Current concepts and caveats. J. Leukoc. Biol. 2015, 98, 557–564.
- Biswas, S.; Thakur, V.; Kaur, P.; Khan, A.; Kulshrestha, S.; Kumar, P. Blood clots in COVID-19 patients: Simplifying the curious mystery. Med. Hypotheses 2021, 146, 110371.
- Tuculeanu, G.; Barbu, E.C.; Lazar, M.; Chitu-Tisu, C.E.; Moisa, E.; Negoita, S.I.; Ion, D.A. Coagulation Disorders in Sepsis and COVID-19—Two Sides of the Same Coin? A Review of Inflammation–Coagulation Crosstalk in Bacterial Sepsis and COVID-19. J. Clin. Med. 2023, 12, 601.
- Bkaily, G.; Jacques, D. Morphological and Functional Remodeling of Vascular Endothelium in Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24, 1998.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278.
- Xu, S.-W.; Ilyas, I.; Weng, J.-P. Endothelial dysfunction in COVID-19: An overview of evidence, biomarkers, mechanisms and potential therapies. Acta Pharmacol. Sin. 2023, 44, 695–709.
- Chen, X.; Zhao, B.; Qu, Y.; Chen, Y.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.; Liu, Y.; Yang, B.; et al. Detectable Serum Severe Acute Respiratory Syndrome Coronavirus 2 Viral Load (RNAemia) Is Closely Correlated with Drastically Elevated Interleukin 6 Level in Critically Ill Patients with Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 1937–1942.
- Kaur, S.; Tripathi, D.M.; Yadav, A. The Enigma of Endothelium in COVID-19. Front. Physiol. 2020, 11, 989.
- Mosleh, W.; Chen, K.; Pfau, S.E.; Vashist, A. Endotheliitis and Endothelial Dysfunction in Patients with COVID-19: Its Role in Thrombosis and Adverse Outcomes. J. Clin. Med. 2020, 9, 1862.
- Mojzisch, A.; Brehm, M.A. The Manifold Cellular Functions of von Willebrand Factor. Cells 2021, 10, 92351.
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56, 2001634.
- Eurelings, L.E.M.; Miedema, J.R.; Dalm, V.; van Daele, P.L.A.; van Hagen, P.M.; van Laar, J.A.M.; Dik, W.A. Sensitivity and specificity of serum soluble interleukin-2 receptor for diagnosing sarcoidosis in a population of patients suspected of sarcoidosis. PLoS ONE 2019, 14, e0223897.
- Marini, J.J.; Gattinoni, L. Management of COVID-19 Respiratory Distress. JAMA 2020, 323, 2329–2330.
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hekimian, G.; et al. Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology 2021, 298, E70–E80.
- Simpson, R.; Mendis, D. A Corpus-Based Study of Idioms in Academic Speech. TESOL Q. 2003, 37, 419–441.
- Cambridge University Press & Assessment. Everything but the Kitchen Sink. Available online: https://dictionary.cambridge.org/us/dictionary/english/everything-but-the-kitchen-sink (accessed on 15 May 2023).
- Roche, J.A.; Roche, R. A hypothesized role for dysregulated bradykinin signaling in COVID-19 respiratory complications. FASEB J. 2020, 34, 7265–7269.
- Wilczynski, S.A.; Wenceslau, C.F.; McCarthy, C.G.; Webb, R.C. A Cytokine/Bradykinin Storm Comparison: What Is the Relationship Between Hypertension and COVID-19? Am. J. Hypertens. 2021, 34, 304–306.
- McCarthy, C.G.; Wilczynski, S.; Wenceslau, C.F.; Webb, R.C. A new storm on the horizon in COVID-19: Bradykinin-induced vascular complications. Vasc. Pharmacol. 2021, 137, 106826.
- Garvin, M.R.; Alvarez, C.; Miller, J.I.; Prates, E.T.; Walker, A.M.; Amos, B.K.; Mast, A.E.; Justice, A.; Aronow, B.; Jacobson, D. A mechanistic model and therapeutic interventions for COVID-19 involving a RAS-mediated bradykinin storm. eLife 2020, 9, e59177.
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37.
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256.
- Jiang, Y.; Rubin, L.; Peng, T.; Liu, L.; Xing, X.; Lazarovici, P.; Zheng, W. Cytokine storm in COVID-19: From viral infection to immune responses, diagnosis and therapy. Int. J. Biol. Sci. 2022, 18, 459–472.
- Rabaan, A.A.; Tirupathi, R.; Sule, A.A.; Aldali, J.; Mutair, A.A.; Alhumaid, S.; Muzaheed; Gupta, N.; Koritala, T.; Adhikari, R.; et al. Viral Dynamics and Real-Time RT-PCR Ct Values Correlation with Disease Severity in COVID-19. Diagnostics 2021, 11, 1091.
- Abdulrahman, A.; Mallah, S.I.; Alqahtani, M. COVID-19 viral load not associated with disease severity: Findings from a retrospective cohort study. BMC Infect. Dis. 2021, 21, 688.
- Hasanoglu, I.; Korukluoglu, G.; Asilturk, D.; Cosgun, Y.; Kalem, A.K.; Altas, A.B.; Kayaaslan, B.; Eser, F.; Kuzucu, E.A.; Guner, R. Higher viral loads in asymptomatic COVID-19 patients might be the invisible part of the iceberg. Infection 2021, 49, 117–126.
- Kim, Y.; Cheon, S.; Jeong, H.; Park, U.; Ha, N.-Y.; Lee, J.; Sohn, K.M.; Kim, Y.-S.; Cho, N.-H. Differential Association of Viral Dynamics with Disease Severity Depending on Patients’ Age Group in COVID-19. Front. Microbiol. 2021, 12, 712260.
- Dorward, D.A.; Russell, C.D.; Um, I.H.; Elshani, M.; Armstrong, S.D.; Penrice-Randal, R.; Millar, T.; Lerpiniere, C.E.B.; Tagliavini, G.; Hartley, C.S.; et al. Tissue-Specific Immunopathology in Fatal COVID-19. Am. J. Respir. Crit. Care Med. 2020, 203, 192–201.
- World Health Organization (WHO). WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 15 May 2023).
- World Health Organization (WHO). Coronavirus Disease (COVID-19): Post COVID-19 Condition. Available online: https://www.who.int/news-room/questions-and-answers/item/coronavirus-disease-(COVID-19)-post-COVID-19-condition (accessed on 15 May 2023).
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Marx, W.; O’Neil, A.; Athan, E.; Walder, K.; Berk, M.; Olive, L.; Carvalho, A.F.; et al. The cytokine storms of COVID-19, H1N1 influenza, CRS and MAS compared. Can one sized treatment fit all? Cytokine 2021, 144, 155593.
- Fung, T.S.; Liu, D.X. Similarities and Dissimilarities of COVID-19 and Other Coronavirus Diseases. Annu. Rev. Microbiol. 2021, 75, 19–47.
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733.
- Bo, Y.; Guo, C.; Lin, C.; Zeng, Y.; Li, H.B.; Zhang, Y.; Hossain, M.S.; Chan, J.W.M.; Yeung, D.W.; Kwok, K.O.; et al. Effectiveness of non-pharmaceutical interventions on COVID-19 transmission in 190 countries from 23 January to 13 April 2020. Int. J. Infect. Dis. 2021, 102, 247–253.
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580.
- Murphy, K.; Weaver, C. Janeway’s Immunobiology; Garland Science: New York, NY, USA, 2016.
- Kasuga, Y.; Zhu, B.; Jang, K.J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736.
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880.