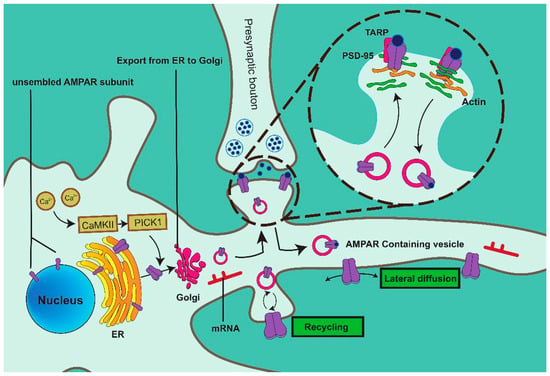
突触可塑性增强或减少神经元之间的连接,影响学习和记忆。突触后 AMPAR 介导谷氨酸能神经元中 90% 以上的快速兴奋性突触传递。AMPAR的数量和亚基组成是突触可塑性和整个神经网络形成的基础。因此,AMPARs在突触后膜的插入和功能化已成为与中枢神经系统神经回路形成和信息处理相关的核心问题。
1. 引言
大脑的正常运作取决于神经元之间通过突触(包括兴奋性和抑制性突触)在神经元之间稳定地传递信息。α-氨基-3-羟基-5-甲基-4-异噁唑吁噁丙酸受体(AMPAR)和N-甲基-D-天冬氨酸受体(NMDAR)是两种通常参与兴奋性突触活动的离子型谷氨酸受体,在大脑神经元的形成和突触可塑性中起着至关重要的作用[
1]。在突触发生过程中,亚基组成的调节以及 AMPAR 和 NMDAR 的相对丰度被认为是建立功能成熟突触的关键。
大量研究表明,突触后膜上同种型和AMPAR数量的精确调节是突触传递和突触可塑性的关键,这有助于形成精确的神经回路[
2,3,4]。AMPAR 聚集、转运和表达缺陷是神经退行性疾病和神经发育障碍(如阿尔茨海默病 (AD)、精神分裂症 (SCZ)、抑郁症和自闭症)的根本原因。
AMPAR是由四个亚基组合而成的四聚体,具有不同的化学计量,具有不同的通道特征,并由四个不同的基因
Gria1-4编码。成年海马体中的AMPAR通常由GluA1/2或GluA2/3组成[
5,6]。这四个亚基是高度同源的,主要在C端结构域上有所不同,这决定了它们在AMPAR运输过程中与不同蛋白质的结合。AMPAR转运过程依赖于PDZ结构域的不同特异性结合,使不同的蛋白质结合效应在下一步中发生。例如,GluA1 的 C 端结构域包含 I 型 PDZ 配体并直接与 SAP97 相互作用,而 GluA2 的 C 末端结构域包含 II 型 PDZ 配体并直接与 PICK1 和 GRIP1 相互作用 [
7]。
AMPAR在内质网(ER)中合成,随后沿着囊泡内的肌动蛋白丝传播到胞吐作用位点。AMPAR通过融合过程释放并插入膜中(图1)。AMPAR在突触膜上的表达是一个高度动态的过程,因为AMPAR在细胞质和质膜之间不断循环。
兴奋性突触的突触强度主要受AMPARs的表达和活性的调控,AMPAR的运输是一个连续的循环过程。钙2+磷酸化并激活 Ca2+/钙调蛋白依赖性调节激酶 II (CaMKII),促进 CaMKII-PICK1 复合物的形成。PICK1 通过其 PDZ 结构域与 GluA2 的 C 端结构域结合,并刺激 GluA2 转运到 ER 膜。AMPARs在被高尔基体(GA)修饰后以囊泡的形式穿过细胞质。一部分AMPAR直接插入突触膜,而其他AMPAR首先通过胞吐作用作用在非突触位点起作用,随后通过横向扩散移动到突触。含有AMPAR的囊泡沿着肌动蛋白丝运输到突触后密度(PSD)。跨膜 AMPAR 相关蛋白 (TARP) 优先结合 AMPAR 四聚体,随后通过其细胞质尾部 (CT) 结合 PSD-95,这是一种促进 AMPAR 募集和定位到突触的机制,确保 AMPAR 在正确的位置发挥作用。
长期增强(LTP)和长期抑制(LTD)是哺乳动物神经元中持久突触可塑性的两种主要形式。大多数关于AMPARs转运机制的研究得出结论,LTP和LTD依赖于AMPARs的胞吐作用和内吞作用[
4,8,9]。在LTP诱导过程中,AMPAR的胞吐作用增加,而增强的内吞作用可导致LTD[
4]。充分的证据表明,LTP和LTD是参与学习、记忆和认知的两种主要分子机制,它们受到AMPAR功能破坏或运输的损害[
10]。
淀粉样蛋白-β(Aβ)的细胞外聚集是AD发病机制中的关键事件,细胞外Aβ水平的调节直接影响疾病的发病。使用定量放射自显影技术,研究表明,与健康受试者相比,AD患者CA1区域的谷氨酸代谢结合和AMPA结合显著降低[
15]。也有报道称,中枢神经系统中的AMPA信号传导介导了谷氨酸能突触的快速传递,并且含有GluA1的突触在AD的早期阶段受损[
16]。
2. AMPAR囊泡形成
2.1. 拉布11
已经证明,AMPARs通过网格介导的内吞作用进入细胞,并储存在Rab11内体区室中。AMPAR在Rab11中沿树突状轴内的微管轨道循环内体运输,去除这些内体会导致AMPAR和PSD-95显著减少[
25]。在LTP期间激活后,AMPAR通过Rab11介导的内体循环返回突触[
26]。
++Rab11属于Rab GTPase家族,存在于各种细胞内膜结合区室的细胞质表面,并调节真核细胞细胞内膜运输的所有阶段,包括囊泡形成、运动、对接和融合[
24]。Rab11是进化上保守且广泛分布的Rab GTP酶亚家族,由三个成员组成:Rab11a、Rab11b和Rab25[
27]。
迄今为止发现的 Rab11 效应蛋白包括 Rab11 相互作用蛋白 (FIP) 家族、Rab11BP、肌球蛋白-Vb、磷酸肌醇 4-激酶 β 和 Sec15。FIP 是 Rab11 驱动的回收内体的重要效应子,提供功能和调控多样性。根据C端Rab11结合结构域,FIP可分为两类:I类(Rab11FIP1/2/5)具有N端C2结构域,II类具有EF-hand结构域[
28]。I类FIP的N端C2结构域靶向Rab11-FIP复合物[
24]。FIP2 作为 LTP 期间 AMPAR 膜运输的负调节因子,与 GluA1 高度共定位。FIP2 包含三个不同的结构域:N 端磷脂结合 C2 结构域、C 端 Rab11 结合结构域 (RBD) 和中央肌球蛋白 Vb 结合结构域 (MBD)。FIP2与GluA1相互作用不需要Rab11结合,FIP2-GluA1解离与Rab11结合无关,这可以防止含有AMPAR的GluA1在没有神经元刺激的情况下到达突触膜[
29]。
2.2. 卡MKII
研究表明,内质网的 GluA2 输出也受 Ca 控制
2+/钙调蛋白依赖性调节激酶 II (CaMKII)。Ca的发布
2+来自 ER 触发介导 GluA2 退出的信号级联(
图 1)。在LTD诱导后,抑制Caa
2+中等大小的棘神经元中内质网的外排阻止了含有GluA2的COPII囊泡离开内质网,从而阻止了GluA2向质膜的运输[
34]。
在LTP过程中,谷氨酸结合刺激NMDAR的活化,以及随后Ca的流入
2+激活CaMKII。CaMKII介导的Stg的C端PDZ结合域磷酸化在其C端尾部产生高度负电荷,排斥膜脂质并使Stg和PSD-95结合,增加AMPAR在突触处的定位[
35]。
3. AMPAR囊泡贩运
3.1. 地图
在内质网中形成后,AMPAR囊泡沿着细胞骨架蛋白(包括肌动蛋白和微管蛋白)移动到细胞膜。微管是细胞骨架的主要成分之一,参与维持突触结构和突触可塑性,是由微管蛋白组成的聚合物[
38],在AMPAR运输过程中与锚定蛋白结合[
39]。在AMPAR转运过程中,微管相关蛋白(microtubule-associated protein, MAP)可能参与突触可塑性的调节[
40]。MAP1A 和 MAP1B 是远亲蛋白,最初合成为多肽,随后裂解成重链 (MAP1B-HC/MAP1A-HC) 和两条轻链 (MAP1B-LC/MAP1A-LC)。MAP1B 轻链 LC1 可以结合 MAP1A 和 MAP1B 重链。MAP1A轻链LC2主要结合MAP1A-HC;然而,它也可以结合MAP1B-HC[
41]。MAP1B被认为参与囊泡融合和突触前结构[
42],LC1已被证明通过GRIP1与AMPAR相互作用。GRIP1提供了一个固定支架,将AMPAR连接到微管,并限制它们进入突触膜[
39]。MAP1A在成熟神经元中表达,MAP1B在未成熟神经元中表达。MAP1A已被证明通过外源性C末端片段结合PSD-95和F-肌动蛋白,从而将谷氨酸受体锚定在细胞骨架上[
43]。
3.2. 肌动蛋白
F-肌动蛋白的丰度使突触具有高度动态性和可变性[
45]。RIL是一种肌动蛋白细胞骨架相关蛋白,在N端包含PDZ结构域,在C端包含LIM结构域(Lin-11、Isl1、MEC-3)[
46]。RIL 与 GluA1 的 C 末端和微丝结合,将 α-actinin-2 与 GluA1 交联并促进 AMPAR 与肌动蛋白的结合。因此,RIL 介导内体 GluA1 和肌动蛋白丝之间的相互作用。含有AMPAR的囊泡通过肌球蛋白-Vb沿着肌动蛋白微丝主动转运至质膜,表明肌球蛋白-V在AMPAR转运中起着至关重要的作用[
47]。各种肌球蛋白亚型参与 AMPAR 运输。肌球蛋白-Va和Rab11参与GluA1从树突状轴到脊柱头部的短期运输[
48,49]。
4. AMPAR锚定在突触后位点
含有AMPAR的囊泡沿着肌动蛋白丝转运到突触后密度(postsynaptic density, PSD),PSD是一个高度组织的支架蛋白网络,由膜受体、信号分子和细胞骨架成分组成[
54]。
PSD 是一种位于神经元突触后膜的独特结构,由突触后密度蛋白 95 (PSD-95;也称为突触相关蛋白 90、SAP-90)、突触后密度蛋白 93 (PSD-93)、突触相关蛋白 97 (SAP-97) 和突触相关蛋白 102 (SAP-102) 组成。这些蛋白属于膜相关鸟苷酸激酶(MAGUK)蛋白家族,该蛋白家族在N端包含3个PSD-95/Dlg/ZO-1(PDZ)结构域,在C端包含Src同源3(SH3)和无活性鸟苷酸激酶(GK)结构域[
55]。在这三个结构域中,PDZ结构域是最重要的蛋白质-蛋白质相互作用结构域[
56],其一般功能是将蛋白质锚定在适当的细胞区室中,从而促进蛋白质支架、信号传导和转运[
57]。PSD-95 是 PSD 中含量最丰富的蛋白质之一。尽管其本身缺乏蛋白活性,但其PDZ结构域通过TARP与AMPAR的相互作用可以有效控制PSD中AMPAR的数量,并介导其在膜上的定位[
54,58]。
4.1. 观星
Stargazin(Stg)是钙离子通道的电压依赖性γ-2亚基,也是TARP蛋白家族中特征最明显的成员,该家族由跨膜蛋白组成,可稳定AMPAR的横向扩散和突触定位[
59]。Stg的C末端的PDZ配体与PSD-95相互作用,并有效地稳定AMPAR在神经元膜上的定位[
60]。事实上,Stg的细胞质尾部是AMPAR运输和定位的关键结构域,而其胞外结构域负责控制通道特性和塑造突触反应[
61]。
4.2. SAP-97的
AMPARs的另一种相互作用蛋白是SAP-97,它参与早期分泌途径以促进AMPAR的成熟[
66],而PSD-95则介导成熟突触的功能[
67]。SAP-97 与 PKA 锚定分子 AKAP-79 相互作用。此外,SAP-97 还通过 PDZ 结构域与 GluA1 结合,这导致 AKAP-79 募集到 GluA1 亚基,从而增强 Ser
845GluA1磷酸化并调节LTP和记忆保留过程[
68]。
MAGUK家族成员通过PDZ域与AMPAR进行交互。SAP-97直接与AMPAR的GluA1亚基结合,而PSD-95需要Stg与AMPAR相互作用[
72]。此外,PSD-95 N末端的棕榈酰化对于AMPAR靶向很重要,而SAP-97介导的AMPAR靶向依赖于位于SH3和GK结构域之间的可变剪接区域(I3结构域)[
73]。此外,SAP-97 N末端的I27结构域负责挽救AMPAR介导的PSD-95敲低诱导的EPSC水平降低[
74]。SAP-97 的所有域似乎都参与了 AMPAR 贩运。
4.3. PKA的
蛋白激酶 A (PKA) 和 C (PKC) 是由 G 蛋白偶联受体介导的信号通路的下游效应子。PKA主要参与以cAMP为次级信使的信号转导级联,而PKC则由磷脂酰肌醇信号通路中产生的二酰基甘油(DAG)激活。PKA 结合并磷酸化 Ser
845位点在GluA1中,促进GluA1在突触膜上的磷酸化,并特异性地增加突触后膜上的AMPAR表达[
78]。然而,并非所有 LTP 都是通过 PKA 介导的 GluA1 磷酸化诱导的,PKA 的磷酸化也不会导致所有 AMPAR 类型的表达增加。缺乏 GluA2 的 AMPAR 转化为钙渗透性 (CP) AMPAR,而含有 GluA2 的 AMPAR 转化为钙不渗透 (CI) AMPAR。LTP可分为早期LTP(E-LTP)和晚期LTP(L-LTP),前者对PKA和蛋白质合成不敏感,后者则相反。AMPAR插入突触后膜的增加导致E-LTP的诱导,这需要CaMKII的激活。E-LTP 可由一次 theta 突发刺激 (TBS) 触发,该刺激可通过调节 Ca 诱导 cAMP 的产生
2+-敏感腺苷酸环化酶。随着 cAMP 表达的增加,L-LTP 增强,cAMP 是随后激活 PKA 的第二个信使。PKA 通过 A 激酶锚定蛋白 (AKAP) 的锚定协调 CP-AMPAR 在突触处的掺入,并调节 GluA1 在 Ser 位点的磷酸化
845,这导致CP-AMPAR插入质膜(
图2)[79,80,81]。
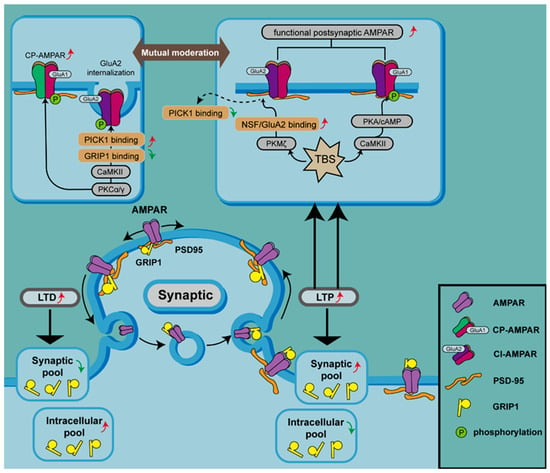
图2.激酶通过磷酸化和细胞内 AMPAR 回收池调节 AMPAR 表达。
AMPAR 的 GluA1 亚基可被 CaMKII、PKC 和 PKA 磷酸化,这有助于在 LTP 期间增加突触膜上 AMPAR 的水平。PKCα/γ 可以磷酸化 GluA2,从而破坏 GRIP1 与 GluA2 的结合并增加 GluA2 与 PICK1 的相互作用,导致含有 GluA2 的 AMPAR 内化和突触膜处 CP-AMPAR 的增加。相反,PKMζ 参与 LTP,因为 PKMζ 通过破坏 NSF/GluA2 结合导致 PICK1 与含 GluA2 的 AMAPR 解离。TBS 可以诱导 cAMP 的产生,从而激活 PKA 并启动 PKA 锚定到 AKAP,协调 CP-AMPAR 在突触处的掺入并调节 GluA1 在 Ser 处的磷酸化845(顶部面板)。
4.4. PKC 和 GRIP 1/2
PKC 是参与上述过程的另一种蛋白激酶,通过诱导 LTP 激活并在 Ser 位点磷酸化 GluA1
831和 Ser
818和 GluA2 在 Ser
880 [
[84]PKC还启动CaMKII的自磷酸化[
85]。研究表明,Ser 的磷酸化
818, Ser
831, and Ser
845 facilitates the insertion of GluA1 into the synaptic membrane, while TBS enhances this phosphorylation at Ser
818 and Ser
831. Of note, among the various candidate kinases and mutant fusion proteins, only PKC phosphorylates Ser
818. Phosphorylation at Ser
818 is critical for LTP by modifying binding to other proteins involved in AMPAR trafficking, such as 4.1N, AP2, and PIP3 [
86].
Activation of one isoform of PKC, PKCα, acts as a trigger for GluA1 phosphorylation at ser
818 and subsequent insertion of CP-AMPARs into the synaptic membrane [
87,
88]. Activation of PKCγ, another isoform of PKC, results in phosphorylation of GluA2 at Ser
880, which disrupts its interaction with synaptic anchoring protein ABP/GRIP, leading to GluA2 internalization [
34] and promotion of CP-AMPAR expression on synaptic membranes (
Figure 2) [
89].
Additionally, it has been found that although AKAPs often bind with PKA to regulate AMPAR trafficking, certain AKAPs, such as AKAP79 [
92], AKAP12 [
93], and AKAP149 [
94], can also regulate PKC. The AKAP79 that functions in PKA phosphorylation of GluA1 was shown to have clearly characterized interactions with the catalytic core of PKC [
92,
95], binds and inhibits the conserved catalytic core of PKCbetaII, and coordinates the subcellular localization of PKC at the postsynaptic site of neurons.
4.5. EPH41L1
Erythrocyte membrane protein band 4.1-like 1 (EPH41L1), also called protein 4.1N, is the third discovered cytoskeletal protein 4.1 superfamily member, which links transmembrane proteins to the actin cytoskeleton [
100]. There exist three highly conserved domains in the protein 4.1 superfamily: the FERM (4.1-ezrinradixinmoesin) domain, the SAB (spectrin-actin binding) domain, and the CTD (C-terminal domain) [
101]. Unlike other 4.1 homologs (4.1B, 4.1G, and 4.1R), the SAB domain of 4.1N does not combine with spectrin or actin. Instead, it interacts with different molecules and other types of proteins through its CTD and FERM domains, which play important roles in modifying synaptic plasticity and synaptic transmission.
5. AMPAR Vesicle Fusion
5.1. SNARE Complex
LTP induction requires NMDAR activation, which promotes calcium influx and subsequent SNARE-dependent membrane fusion of AMPAR-containing endosomes. The SNARE complex is typically composed of SNARE proteins localized in vesicles (v-SNARE) and target membranes (t-SNARE), which are essential for the membrane fusion process [
106]. As AMPAR-containing vesicles move along actin filaments to the locality of the plasma membrane, the v-SNARE protein synaptobrevin-2 (Syb-2) on the vesicle interacts with two t-SNARE proteins, syntaxin-3 (Stx-3) and SNAP-47, on the plasma membrane to form a SNARE complex; thus, vesicles containing AMPARs are anchored to the synaptic exocytosis site [
105]. Initially, Stx-3 binds to the SM (Sec1/Munc18-like) protein Munc-18, which organizes the assembly of v-SNAREs with t-SNAREs [
107]. After assembly, SNARE complexes bind to Complexin, producing an activated but frozen state [
107]. Upon NMDAR activation and subsequent calcium influx, the calcium sensor Synaptotagmin-2 (syt2) on AMPAR-containing vesicles receives Ca
2+ signals and removes Complexin, promoting membrane fusion [
108] and the insertion of AMPARs into the synaptic membrane.
NSF is a hexamer ATPase that participates in the recycling of SNARE complexes. NSF promotes the disassembly of the SNARE complex and accelerates its recycling by hydrolyzing ATP to increase the abundance of AMPARs at the membrane. In addition, loss of NSF results in decreased expression of GluA1–3 at the membrane. Since NSF binds directly to the C-terminal domain of GluA2 and does not interact with GluA1 or GluA3, decreases in the expression of GluA1–3 may manifest as a reduction in GluA2 at the membrane [
110].
5.2. AP-2
During LTD, AMPARs are removed from the cell surface by endocytosis, after which a pool of AMPARs in the endosomal recycling compartment can be transported to the dendritic spine and spread laterally to the PSD [
115].
The adaptor protein 2 complex (AP-2 complex) is mainly involved in the induction of AMPAR internalization; the μ2 subunit of AP-2 binds directly to a basic motif with high affinity, which is present in the cytoplasmic tail (CT) of GluA1–3 and in the presynaptic vesicle protein Synaptotagmin 1 [
116]. It is noteworthy that the binding site for AP-2 in GluA2 overlaps with that for NSF [
117], indicating that AP-2 may compete with NSF for the binding of GluA2. The binding of NSF to GluA2 enhances AMPAR expression at the synaptic surface by facilitating the delivery of GluA2 to the postsynaptic membrane [
111], while AP-2 promotes the endocytosis of AMPARs.
AMPARs, synthesized in the ER, are glutamate ionotropic receptors located in the PSD. In the absence of neuronal stimulation, FIP2 binds to the GluA1 subunit of AMPARs to prevent entry into the synapse [
29]. Following LTP induction, glutamate binds to and activates NMDARs [
119], which results in Ca
2+ entering postsynaptic cells and activating CaMKII to phosphorylate GluA1 and Stg (γ
2). Vesicles containing phosphorylated GluA1 subunits bind to the Rab11 adapter complex, and Ca
2+-sensitive Myosin-Va carries this complex and AMPAR-containing vesicles along the actin filaments to the dendritic spine head. Ca
2+-activated Myosin-Vb transports AMPARs to the exocytosis site along actin filaments [
120,
121].
6. The Actin Cytoskeleton That Facilitates AMPAR Trafficking
AMPAR trafficking involves multiple dynamic processes of vesicle cycling, all of which involve the actin cytoskeleton, which is highly dynamic and regulated. Actin dynamics generate forces that manipulate the membrane during vesicle biogenesis and are also used to propel vesicles through the cytoplasm to their destinations. Actin and actin-binding proteins (ABPs) are highly enriched at the postsynaptic density (PSD), where actin cytoskeleton dynamics maintain dendritic spine morphology during neuronal development and undergo expansion and contraction during synaptic activity changes [
124].
Furthermore, some other proteins involved in AMPAR transport have also been found to have an inseparable relationship with the actin cytoskeleton. For example, the actin-regulatory protein cortactin acts as an intracellular sorting mediator for AMPARs. Disruption of the binding between GluA2 and cortactin in neurons results in the targeting of GluA2/A3 receptors to lysosomes and subsequent degradation, leading to the loss of surface and synaptic GluA2 under basal conditions and blocking subsequent LTD expression [
125].
重塑脊柱肌动蛋白网络的第一步是解离肌动蛋白丝,肌动蛋白丝通常由未磷酸化的CamKIIβ交联[
127]。CamKIIβ 通过将两根细丝(或 PSD 处的细丝和蛋白质)结合在一起来防止其他 ABP 的结合,从而稳定肌动蛋白网络。在 LTP 诱导过程中,CaMKIIβ 被钙/钙调蛋白结合和 CaMKIIα 磷酸化激活。然后,CaMKIIβ 与 g-肌动蛋白解离,使 g-肌动蛋白单体可用于细胞骨架网络生长。此外,CaMKIIβ和α-actinin4(另一种与肌动蛋白和PSD蛋白结合的交联蛋白)的解离允许其他ABP结合肌动蛋白[
128]。这些结果表明,CamKII可能在脊柱形态和突触可塑性的调控中发挥更复杂的作用。AMPAR组装发生在内质网中,并通过囊泡转运到细胞表面。研究表明,突触强度的增加与内质网的稳定锚定相关[
129]。在哺乳动物的中央突触中,在任何给定时间点,只有一小部分棘含有内质网(15–50%[
130,131]),但这种树突状内质网具有高度动态性,并且随着时间的推移会瞬时地探索大多数棘[
129]。
7. AMPAR贩运神经系统疾病
由于AMPAR在神经系统中的关键作用,大量研究表明,靶向AMPAR可以通过利用各种AMPAR激活剂或拮抗剂来影响相关神经系统疾病的发展。然而,这些研究大多只关注直接补充或拮抗AMPAR和观察疾病进展,而没有进一步研究AMPAR转运在各种疾病中的具体过程和机制。这种对AMPAR的简单调节可能不会产生持久的效果。然而,随着对AMPAR在疾病中的研究不断深入,对AMPAR在疾病中的转运机制逐渐有了更多的解释,这有助于我们更详细地了解疾病的进展,并开发更有效的治疗或预防方法。
AMPAR相关的囊泡形成和转运过程极易感染疾病。Rab11 作为内吞作用和细胞内囊泡循环的重要蛋白质,在各种神经系统疾病中起着至关重要的作用,尤其是与蛋白质病理聚集相关的疾病。在亨廷顿病(HD)中,亨廷顿蛋白(HTT)调节Rab11依赖性转运,Rab11依赖性转运参与维持树突棘的大小[
136]。突变HTT片段会破坏Rab11介导的内吞转运并损害GluA1的膜定位,而
Rab11的过表达可恢复果蝇模型中的HD相关表型[
137]。早期阿尔茨海默病(Early Alzheimer's disease, AD)在内吞周期中表现出功能障碍,其对细胞内循环系统的损害与Rab11密切相关[
138]。研究表明,BIN1是迟发性阿尔茨海默病(late-onset Alzheimer's disease, LOAD)的危险因素,与Rab11共定位,与AMPAR亚基GluA1形成复合物,有助于维持阿尔茨海默病的正常脊柱形态和AMPAR介导的突触传递[
139]。有趣的是,在脑缺血和再灌注 (OGD/R) 期间,只有含有 GluA1 亚基的 AMPAR 与 Rab11 阳性内体共定位,而 GluA2 亚基不存在于 Rab11 阳性内体中。这可能表明GluA1和GluA2在OGD/R过程中的差异膜表面回收机制[
140]。
Surprisingly, TARPs, such as stargazin, which stabilizes AMPARs, have been found to directly participate in disease processes in various diseases. Brain quantitative proteomic analysis of AD models showed that stargazin gradually disappears from AMPAR complex sets with increasing Aβ levels, accompanied by impaired glutamatergic synaptic plasticity. Supplementing with stargazin can restore impaired excitatory synaptic transmission in AD [
144]. In patients with intellectual disabilities, a stargazin V143L missense mutation disrupts the interaction between stargazin and AMPAR, resulting in AMPAR transport defects, synaptic dysfunction, and the manifestation of cognitive and social behavioral impairments associated with intellectual disability [
145].
HTT mutations in HD have been found to weaken the interaction between HTT and stargazin-PSD95, accompanied by a surface diffusion imbalance of AMPARs [
146]. Stargazin exhibits higher S-nitrosylation in the ventromedial prefrontal cortex (vmPFC) of anxious mice. Injection of an exogenous stargazin (C302S) mutant can alleviate S-nitrosylation, rescue GluA1 and AMPAR-mediated synaptic transmission, and improve anxiety-like behaviors [
147].
OGD/R-induced impairment of AMPAR trafficking was shown to be PICK1-dependent [
150]. Disrupting the interaction between PICK1-GluA2 and the c-terminal GluA2 peptide (EVKI) can effectively attenuate OGD-induced GluA2 internalization [
150]. Additionally, other research has suggested that AP is also involved in this process, as AP interacts with PICK1 to mediate the OGD-induced endocytosis and degradation of GluA2 [
151]. The interaction between PICK1 and GluA2 also plays a critical role in neuropathic pain, affecting hyperalgesia in sciatic nerve constriction injuries [
152]. Mutations in
RAB39B, associated with intellectual disability and epilepsy, can also bind to PICK1 to regulate the AMPAR subtype ratio and alter synaptic activity [
153].
HTT controls the transport of AMPARs containing GluA2 along microtubules in dendrites through the motor protein kinesin 5 (KIF5). In HD models, the GluA2/KIF5/HAP1 complex is disrupted, leading to loss of microtubule binding ability and impaired movement of vesicles containing AMPARs along microtubules [
158]. Additionally, HTT affects actin dynamics through the LIMK-cofilin pathway, playing a role in recruiting AMPAR to active synapses. Loss of
HTT results in a disconnection between spine structure and synaptic function, potentially contributing to the development of HD symptoms [
126]. Enrichment analysis of hippocampal LTP protein components related to AD has highlighted the critical role of the actin cytoskeleton pathway in hippocampal LTP [
144].
8. AMPARs PTM
In addition to the subunit-specific protein interactions, AMPARs also undergo many post-translational modifications (PTM) that contribute to the regulation of synaptic activity and plasticity (
Figure 3). Among the three current major PTMs of AMPARs, phosphorylation and palmitoylation mainly influence receptor channel conductance and intracellular trafficking, while ubiquitination serves as the signal for receptor internalization and proteasomal degradation [
4]. Lysine acetylation is a newly described PTM of AMPARs [
159], which represents the only known modification of receptors on stable cell membranes and thus has great research potential.
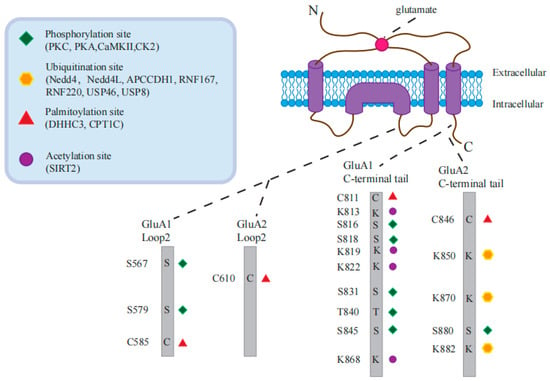
Figure 3. AMPAR post-translational modification sites and related proteins.
8.1. Phosphorylation in AMPAR Trafficking
AMPARs serve as a substrate for various proteins, facilitating the occurrence of synaptic plasticity mechanisms like long-term potentiation (LTP), long-term depression (LTD), and other forms. These proteins, including protein kinase C (PKC), cyclic AMP-dependent protein kinase/protein kinase A (PKA), and calcium/calmodulin-dependent protein kinase II (CaMKII), have been extensively studied by researchers. They interact with the C-terminal region of different AMPAR subunits through various modifications such as phosphorylation and dephosphorylation, thereby regulating the synaptic transport and plasticity of AMPARs.
In particular, phosphorylation of GluA1 at Ser
845 by PKA and at Ser
831 by CaMKII or PKC has been extensively studied. PKA-mediated phosphorylation of GluA1 at Ser
845 has been shown to promote its cell-surface insertion and synaptic retention, whereas dephosphorylation at Ser
845 is related to receptor endocytosis, LTD, and homeostatic scaling-down [
160]. Following the binding of Ca
2+ to CaMKII, auto-phosphorylation occurs to activate CaMKII, which phosphorylates GluA1 at Ser
831, not only promoting the insertion of AMPARs into the postsynaptic membrane but also enhancing channel conductivity and eventually facilitating LTP [
161].
PKC-mediated phosphorylation of GluA1 at Ser
816 and Ser
818 enhances its interaction with 4.1N, which promotes GluA1 insertion into the postsynaptic membrane, consequently strengthening LTP. However, palmitoylation of GluA1 at Cys
811 disrupts its interaction with 4.1N and impairs LTP [
102]. PKC-mediated phosphorylation of GluA2 at Ser
880 interferes with its interaction with GRIP [
162].
8.2. Ubiquitination in AMPAR Trafficking
Nedd4, Nedd4L, APCCdh1, and RNF167 are the four AMPA receptor-specific E3 ubiquitin ligases that have been shown to control synaptic activity in primary neurons. [
166,
167,
168,
169,
170]. It has been reported that only two specific E3 ubiquitin ligases, GluA1 and GluA2, which can ubiquitinate in the C-terminal domain, have been identified [
171]. Nedd4, Nedd4L, and APCCdh1 regulate GluA1 ubiquitination [
166,
167,
168,
169], while RNF167 regulates GluA2 ubiquitination [
170]. The GluA1 subunit is primarily ubiquitinated at the C-terminal, while GluA2 is predominantly ubiquitinated at Lys
870 and Lys
882 in the same region [
172]. In addition, although RNF167 regulates GluA1 and GluA2 expression on the surface of neurons [
170], only its ubiquitination function has been studied, and it is unclear whether these E3 ligases have specific selectivity for these two AMPAR subunits. According to statistics, the researchers have found that 83 of the 600 to 700 E3 ubiquitin ligase genes in the genome are associated with brain disease [
173].
Some evidence exists to support the view that the ubiquitination of GluA1 is an endocytosis signal for AMPARs. For example, the overexpression of
Nedd4-1 in neurons enhances the ubiquitination of GluA1, which results in a decrease in the number of AMPARs at the plasma membrane [
166,
167,
176]. Moreover, the knockdown of
Nedd4-1 increases the rate of GluA1 internalization [
176]. Similarly, overexpression of
USP46 downregulates the ubiquitination of GluA1 and reduces the accumulation of internalized GluA1 in neurons [
177]; therefore, it has been suggested that ubiquitination of AMPARs is necessary for internalization and thus occurs prior. Another view suggests that endocytosis precedes the binding of ubiquitin to AMPAR. For example, inhibition of enzymes critical to membrane fission during endocytosis or of the formation of clathrin-coated pits has the potential to impede the ubiquitination of all subunits of AMPARs in cultured neurons upon exposure to either AMPA or bicuculine [
178].
8.3. Palmitoylation in AMPAR Trafficking
Another form of AMPAR modification of AMPARs is the covalent attachment of the lipid palmitate via thioester bonds at their intracellular cysteine residues. Protein S-palmitoylation, a post-translational modification, resembles phosphorylation in its reversible and labile nature [
182]. The catalysis of this process is facilitated by palmitoyl acyltransferases (PATs) containing conserved Aspartate–Histidine–Histidine–Cysteine (DHHC) motifs, while the reversal is carried out by depalmitoylating enzymes like acyl protein thioesterases and palmitoyl protein thioesterases.
Palmitoylation of GluA takes place in two separate groupings: one found within the second transmembrane domain (TMD2) and another at the C terminus in the juxtamembrane domain near the fourth TMD (TMD4). The palmitoylation process of GluA1 at TMD2 is facilitated by DHHC3, a Golgi-resident PAT. Elevated levels of palmitic acid and DHHC3 cause GluA1 palmitoylation, promoting GluA retention in the Golgi and suppressing its activity-dependent delivery to the plasma membrane [
183], implying that depalmitoylation of AMPARs at the Golgi apparatus would be a signal for their release to the cell surface.
Specifically, depalmitoylation at Cys
585 in GluA1 favors trafficking to the plasma membrane. Carnitine palmitoyltransferase 1C (CPT1C) belongs to the family of carnitine long-chain acyltransferases and is located in the ER of neurons [
184]. It has been demonstrated that CPT1C-mediated enhancement of the cell surface expression of GluA1-containing AMPARs is dependent on the palmitoylable Cys
585 residue [
185], indicating that alterations in the palmitoylation condition of GluA1, which is facilitated by CPT1C, could potentially account for this observed outcome.
In the ER, AMPARs are positioned with their palmitoylation sites facing the cytoplasmic side [
186], and the catalytic domain of CPT1C also faces the cytoplasm; therefore, this topology allows contact between the two protein domains. Thiolipase activity is performed by a catalytic triad of the CPT1C domain that transports the palmitoyl moiety from the substrate (GluA1) to the carnitine molecule, leading to the depalmitoylation of GluA1, which increases the surface expression of AMPARs [
187].
8.4. AMPAR贩运中的乙酰化
乙酰化是蛋白质的一种翻译后修饰,直到近年来才被广泛研究,是酰基辅酶A化合物在乙酰转移酶的作用下与特定残基(通常是赖氨酸)的共价连接。乙酰化发生在组蛋白和非组蛋白上。
最近的一项研究表明,赖氨酸乙酰化是 AMPAR 的一种新的翻译后修饰,已被证明能够延长 AMPAR 的半衰期并稳定细胞膜上的受体修饰,从而在大脑中起着至关重要的作用。AMPAR的乙酰化似乎排斥泛素化。当AMPAR被乙酰化时,不会发生泛素化,这可能是因为当乙酰基重新放置赖氨酸位点时,AMPAR亚基受到保护[
167]。
一项研究[
159]发现,AMPAR在碱性条件下具有高水平的乙酰化,可通过神经元活动有效调节。乙酰化增加导致AMPAR在膜上的表达增加,并在体外和体内增强基本突触传递强度。SIRT2已被确定为AMPAR的脱乙酰酶[
159]。AMPAR的稳态平衡是由于SIRT2对GluA1的C端赖氨酸残基的竞争,导致AMPAR的乙酰化和泛素化的相对权重达到平衡。
8.5. AMPAR PTM之间的串扰
AMPAR的突触转运和递送功能受到不同翻译后修饰的功能相互作用的串扰,以精确调节AMPAR介导的突触信号转导[
4]。最近的一项研究反映了这一结论,其中抑制 GluA1 泛素化导致 Ser 的 PKA 磷酸化水平升高
845,而 PKC/CaMKII 在 Ser 位点磷酸化
831保持不变[
189]。
9. AMPAR PTMs在神经系统疾病中的应用
磷酸化作为研究最广泛的蛋白质修饰之一,在神经系统中已被广泛讨论。对AD的研究表明,Aβ在LTD过程中驱动GluA2中S880残基的磷酸化,GluA2 S880E突变体的表达阻止了LTD驱动的内化,同时也阻断了Aβ诱导的形态和突触抑制。模拟这种AMPAR磷酸化会产生类似于Aβ诱导效应的脊柱形态和突触抑制[192]。在小鼠持续炎症性疼痛模型中检测到 GluR1 膜插入增加和 GluR2 内化增加,并伴有 PKCα 介导的 GluA2 在 Ser 位点磷酸化增加880,脊髓中的PICK1也促进了这一过程[152]。在小鼠神经损伤(SNI)模型中,发现使用PICK1的短肽抑制剂tPD5可以完全缓解神经性疼痛,同时还可以减少脊髓中GluA2 S880的磷酸化,逆转GluA1和GluA2的表面表达,以及神经性疼痛中CP-AMPARs的膜插入[193]。其他研究表明,由于钙/钙调蛋白依赖性蛋白磷酸酶 (PPP3) 去磷酸化活性的丧失,PKCγ 介导的 PKCγ 介导的 AMPAR 的持续磷酸化可能导致神经性疼痛,并且与 GluA1 在 Ser 位点的磷酸化有关831(但不是 Ser845)通过PKCγ而不是PKA或CaMKII[194]。抑制PI3K/AKT通路还阻止了含有GluA1的AMPAR的磷酸化和表面表达,减轻了大鼠模型中的疼痛行为并减弱了中枢敏化,从而缓解了IS诱导的慢性偏头痛[195]。这表明神经性疼痛中存在 GluA1 和 GluA2 的差异磷酸化调节通路,并且通过不同的磷酸化通路调节 CP-AMPAR 的膜表面含量可能至关重要。癫痫的特征是海马神经元过度兴奋和AMPARs过度激活,与AMPAR磷酸化密切相关。Vezatin在癫痫患者中异常表达,与PKA相互作用。维扎汀的敲低可降低磷酸化PKA(pPKA)的水平,从而降低S845位点GluA1的磷酸化,下调GluA1亚基的表面表达,并抑制癫痫小鼠的癫痫发作。这表明癫痫发作的病理机制可能涉及GluA1磷酸化对癫痫发作强度的调节[196]。
This entry is adapted from the peer-reviewed paper 10.3390/ijms25010111