Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Certain clusters of bacteria have been associated with fertility and health, while the outgrowth of several species is potentially correlated with infertility indicators. This constitutes a compelling reason for outlining the external elements that may induce changes in the seminal microbiome composition, like lifestyle factors, gut microbiota, pathologies, prebiotics, and probiotics.
- microbiome
- semen
- sperm
- fertility
- human
- bacteria
- viruses
1. Introduction
Mammalian bodies contain microbial communities that inhabit commensally in diverse tissues. These niches include bacteria, viruses, and fungi and are known as microbiota. The microbiota and the host establish a mutually beneficial relationship, with the host providing favorable habitat conditions for microorganisms, and the microbiota supports good development of the immune and metabolic system [1]. In particular, the human microbiome is restricted to exterior tissues and invaginations like skin, mucosa, gut, or genital tract [2] and is mainly composed of Firmicutes and Bacteroidetes bacterial phyla (92%) [3]. Nevertheless, the niches of microbiota found in the human body are not a stable population but a dynamic set of communities that change over the years and are influenced by factors like the environment, diet, lifestyle, and diseases [4,5].
Although the microbiome of the main human niches has been thoroughly profiled during the last few decades, only a small proportion of studies were dedicated to characterizing the seminal microbiota. The evolution of the microbial profiling technologies over the years have allowed us to define the main populations of bacteria and viruses that inhabit the seminal fluid and their variations due to external factors like sexual life, diseases, or infertility. Many authors have focused on this last topic and found the association between the over-representation of certain species and specific effects related with fertility disorders.
The potential role of semen microbiome in fertility has become a field of special interest, given that infertility constitutes a growing problem that affects 8–12% of couples worldwide, with male-specific factors contributing to 40–50% of those cases [6]. Knowing the mechanisms that determine the influence of seminal microbiota over fertility opens new doors to developing novel approaches and treatments. In this sense, the potential use of beneficial bacterial species through the intake of prebiotics and probiotics is starting to display an interesting potential.
2. Origin of the Seminal Microbiome and General Composition
Originally, it was believed that the microbiota detected in seminal fluid was a footprint of previous or current infections in the urogenital tract and a potential reflection of inadequate cellular immune responses [7,8]. With the application of novel technologies, the idea of a commensal community of seminal microorganisms has arisen since bacterial and virus populations have been detected in samples from patients who are healthy, infected, fertile, or infertile [9,10]. In fact, the slightly basic pH and molecular composition of semen compose a suitable habitat for microorganisms [11].
Therefore, the composition of seminal microbiota results from the contribution of diverse zones and fluids of the urogenital tract, like urine, the urethra, and the coronal sulcus [11]. Also, a contribution of the gut microbiota is possible since there is a strong interaction between the gut microbiome and the regulation of testicular functions, known as the gut–testes axis [12].The testicular and epididymal contribution to the seminal microbiome community has been suggested since an alteration of semen microbiota has been identified after vasectomy [13]. Also, there is an overlap between the microorganism profiles of seminal fluid and urine/testicular samples [13,14,15,16,17]. Nevertheless, these repertoires are only partially shared, so semen harbor species not displayed in first-catch urine samples [18], suggesting a specific microbiome niche.
From the first characterization studies of the human seminal microbiota [19,20], many authors have researched its microbial imprint. Although the results obtained through the years have been heterogeneous and variable, possibly due to differences in methodology and in the selection of the study cohort (see Section 5), a common pattern starts to be elucidated.
The most comprehensively characterized microorganisms in human semen are bacteria. Some specific genera are especially frequent among profiling studies, like Staphylococcus, Streptococcus, Lactobacillus, Corynebacterium, Prevotella, Escherichia, Anaerococcus, Enterococcus, Finegoldia, Peptinophilus, Vogesella [7,8,10,16,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39], and also Gardnerella, Campylobacter, Ureaplasma, Haemophilus, Klebsiella, and Pseudomonas [7,10,20,21,22,24,25,27,30,33,34,35,36,38,39]. Other genera like Acinetobacter, Cutibacterium, Porphyromonas, Chlamydia, Bacillus, Burkholderia, Morganella, Pelomonas, and Proteus were identified [10,20,22,29,30,33,34,35,36,38,39].
The nucleic acid footprint of viruses have also been detected in the human seminal fluid, probably persisting after infections that have been transmitted to the genital tract, especially in cases of viremia [40]. They can be found as free virus particles, attached to molecules on the outside of spermatozoa or inside them. In fact, apart from mature spermatozoa, they can also infect their precursor cells and seminal immune cells [41]. Signs of several viruses from the Flaviviridae family have been persistently detected in semen samples, especially Zika virus (ZKV) [39,40,42]. Different species from the Herpesviridae family, like herpesvirus and cytomegalovirus (HCMV) [39,40,43,44,45,46], and from the Papillomaviridae family [43,44,45,47,48] are also frequent, as well as Parvoviridae like adeno-associated viruses (AAV) [40,44]. The human immunodeficiency virus (HIV) is one of the most cited virus regarding seminal microbiota [49]. Several studies have detected SARS-CoV-2 in semen after COVID-19 infection [50,51].
3. The Semen Microbiome in Fertility Disorders
As mentioned before, the seminal microbiota is involved in the maintenance and regulation of homeostasis and health. Specifically, its potential role in fertility is becoming clear. It is known that infections and inflammatory reactions in the male genital tract are the cause of around 6–10% of infertility cases [49]. In fact, many studies have discovered that fertile and infertile populations displayed different bacterial cohorts in the seminal fluid. Data obtained so far point out that infertility implies a higher richness and diversity of microbial elements, with an increase in alpha diversity (relative to the number of different taxa) [9,13]. The dysbiosis and abundant detection of microorganisms have been related to different indicators of poor fertility status, like seminal ROS, sperm DNA fragmentation, and the disruption of Protamine 1/Protamine 2 (P1/P2) ratio [16,37,52,53]. It is known that abnormal sperm P1/P2 ratios are related with higher DNA fragmentation and also with sperm parameters and fertilization capacity [52]. Moreover, the high presence of specific bacterial genera has been related to fertility disorders. Some examples are Cutibacterium, Rhodopseudomonas [21], Aerococcus [13], Varibaculum, Escherichia [9], Mycoplasma, Ureaplasma [54], or Chlamydia. In particular, U. urealyticum, U. parvum, and M. hominis, which inhabit the male urethra and contaminate semen during ejaculation, are known to play an etiologic role in genital infections and infertility, although their mechanisms have not been discovered yet [54,55]. Furthermore, infection by C. trachomatis has been associated with the induction of sperm apoptosis and impacting fertilizing ability [14,56].
Regarding other species suspected to affect specific sperm parameters, infections with the bacteria Streptococcus, Klebsiella, Ureaplasma parvum, and Chlamydia trachomatis, as well as ZIKV and AAV, have been associated with low rates of progressive and non-progressive motility of sperm cells [32,56,79,80,81,82]. In general, the lipopolysaccharide contained in the cell walls of gram-negative bacteria (like Bacteroidia, Sphingobacteria, Proteobacteria, or Alphaproteobacteria) are suspected to disrupt sperm motility [28]. Low rates of normal morphology in sperm have been observed in samples with the abundance of Staphylococcus aureus, Ureaplasma urealyiticum, HCMV, and human polyomavirus 2 (JCPyV) [34,76,83,84,85], and low sperm counts have been associated with the presence of Staphylococcus, Haemophilus, Klebsiella, Chlamydia trachomatis, and ZIKV [25,32,56,74,76,82]. Patients with azoospermia have shown more abundance of Mycoplasma and Ureaplasma [27], as well as an increase in Bacteroidetes, Firmicutes [86], Proteobacteria, and Actinobacteria [87].
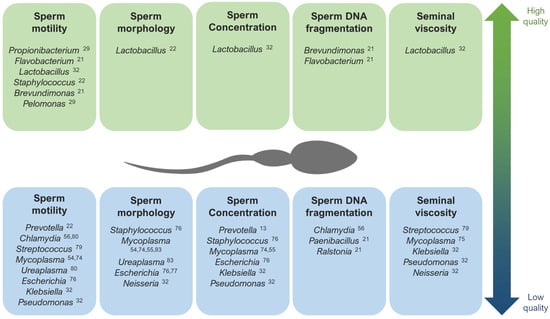
Contrarily, Lactobacillus tends to predominate in the semen microbiota of healthy and fertile men with good quality semen parameters [22,31,32,57,78,88]. Brevundimonas, Staphylococcus, Flavovacterium, and Pelomonas have also been detected in patients with good seminal indicators, like high rates of sperm motility and low DNA fragmentation [21,22,27,29] (Figure 1).
Lastly, when studying the microbiome composition of semen in relation with pregnancy rate, men involved in spontaneous pregnancy loss displayed the presence of Porphyromonas and Campylobacter [89], S. aureus and E. coli [90], as well as a high viral diversity [43]. Contrarily, high rates of success in assisted reproduction techniques (ART) has been associated with Acinerobacter, Lactobacillus Jesenia, and Faecalibacterium [27,89].
4. Influence of External Factors over Sperm Microbiome Composition
Several studies have made direct associations between external factors and the transformation of seminal microbial composition (Figure 2). Commensal microbiota can suffer alterations due to the influence of external factors. One of the most studied cases in the human body is the gut microbiota: many studies have revealed an influence of factors like lifestyle, age, ethnicity, geographical location, body mass index, food, diseases, and treatments over the composition of the microbiota residing in the guts [11]. Taking into account the microbial connection through the gut–testes axis, these factors affecting the gut microbiome may also produce alterations in the seminal microbiota.
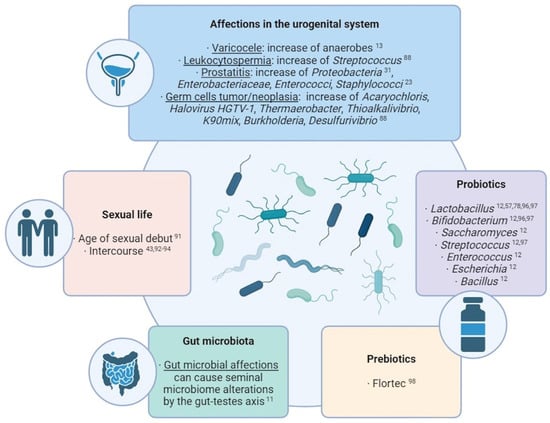
In this sense, alterations in the urogenital system can also produce modifications in its own semen microbiota. For example, an over-representation of anaerobes has been observed in seminal samples of patients with varicocele [13], and Streptococcus-enriched microbial communities are associated with leukocytospermia [88]. In cases of prostatitis, a wide species richness and a higher proportion of Proteobacteria was observed [31], while the growth of Enterobacteriaceae, Enterococci, and Staphylococcus was observed in the semen of chronic prostatitis syndrome patients compared with healthy men [23]. Regarding testicular germ cell tumors and germ cell neoplasia, high levels of Acaryochloris marina, Halovirus HGTV-1, Thermaerobacter marianensis, Thioalkalivibrio sp. K90mix, Burkholderia sp. YI23, and Desulfurivibrio alkaliphilus have been detected in the seminal plasma of the affected patients [88].
5. Technical Considerations in the Study of the Seminal Microbiome
Given the fact that seminal microbiota—and microbiota in general—has a heterogeneous and changing nature, designing a microbial characterization study is a challenging goal since technical variation could contribute to bias in this profiling. These potential sources of variation may include specimen inclusion and exclusion criteria, processing of the samples, accidental contamination, the selected biological fraction, and the chosen technology or platform.
Most of the studies published so far have used whole ejaculate samples for the analysis, regardless of the profiling strategy. Nevertheless, several authors include a centrifugation or wash step prior to the molecular analysis with the cellular fraction. This is frequent for the study of viruses in sperm cells [47,67,82] but can also be conducted in bacteria profiling methods [31,32,33,56,57,88]. Štšepetova et al. studied the bacterial populations in raw ejaculates and processed/washed sperm samples, finding remarkable differences between them [28], indicating potential differences in the microbiota of seminal fluid and associated with cell fraction.
In fact, the use of PCR and qPCR started to gain popularity in the 2000s’ decade, which were frequently used for the detection of Mycoplasma, Ureaplasma and Gardnerella [14,55,56,80], although other studies also included Chlamydia and Neisseria [56,80] and other species of bacteria and viruses to the panel like HPV, Enterococcus, Streptococcus, Staphylococcus, Escherichia, Pseudomonas, Klebsiella, and Lactobacillus [34]. This technique offered a more sensitive detection of different bacterial entities at a molecular level in comparison with culture, as well as a quantification of the sample in the case of using qPCR. Nevertheless, the need for a previous selection of the species of interest to be included in the analysis supposes a serious handicap.
This limitation was overcome with the incorporation of next-generation sequencing (NGS) to the microbiome research field. This technology is usually based on the sequencing of certain hypervariable regions of the 16S rRNA gene of bacteria, which allows the distinction of operational taxonomic units (OTUs) at species level. As metagenomics technology, this approach provides a massive molecular characterization and quantification without a previous selection of species and offers wide possibilities of subsequent bioinformatics analyses, like taxonomic diversity studies (alpha and beta diversity) or clustering. The characterization of semen microbiota through 16S rRNA sequencing has broadened the range of bacterial elements discovered and shed light on aspects like their potential association with health and fertility [9,25,26,34,79,87,89] or their possible inter-relations [22,57]. In addition, other NGS strategies have been applied in the study of seminal microbiota. Some authors have performed bulk RNA-seq over sperm samples and used the reads that were not aligned to the human genome to perform a transcriptome characterization of bacteria and viruses altogether [33,39,88]. Lundy et al. also combined the 16S rRNA sequencing strategy with shotgun metagenomics [13], and Garcia-Segura et al. performed a bacterial profiling full-length 16S rRNA sequencing [21,24].
This entry is adapted from the peer-reviewed paper 10.3390/biology13030150
This entry is offline, you can click here to edit this entry!