Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Infectious Diseases
Antibiotic-related adverse events are common in both adults and children, and knowledge of the factors that favor the development of antibiotic-related adverse events is essential to limit their occurrence and severity. Genetics can condition the development of antibiotic-related adverse events, and the screening of patients with supposed or demonstrated specific genetic mutations may reduce drug-related adverse events.
- antibiotics
- antibiotic prescription
- antibiotic-related adverse events
- genetic variants
- pharmacogenomics
- pharmacokinetics
1. Introduction
Antibiotics have been incredibly effective in enhancing health outcomes in humans. With the introduction of these drugs in clinical practice, many once deadly bacterial infections have been effectively treated in the last 70 years, with a significant reduction in morbidity and mortality [1,2]. However, to maximize and maintain these benefits, the prescription of antibiotics must be carefully considered. The use of these drugs is associated with the development of a number of relevant microbiological and clinical problems that can minimize or cancel the antimicrobial efficacy of the prescribed drugs. The abuse and misuse of antibiotics is the most significant cause of the emergence of microbial resistance to commonly prescribed antibiotics, with a progressive reduction in their clinical efficacy, the reemergence of problems regarding the treatment of some bacterial diseases and the need for the development of new antibacterial agents [3]. Moreover, the administration of antibiotics can be associated with the development of short- and long-term adverse events that may be clinically relevant and lead to the need for medical intervention, hospitalization, admission to the intensive care unit and, although rarely, patient death [4,5].
Antibiotic-related adverse events are common in both adults and children, although they are more common among pediatric patients, probably due to the larger use of these drugs in the first years of life. A study carried out in the USA involving mainly adults showed that, in 2013–2014, about 16% of the Emergency Department (ED) visits for adverse drug events were associated with a previous antibiotic prescription [6]. Of these, about 45%, mainly old people, required hospitalization. In children, studies have found that antibiotic-related adverse events cause approximately half of all medical visits for drug-related medical problems and that about 40% of these involve children aged ≤2 years [7,8].
Knowledge of the factors that favor the development of antibiotic-related adverse events is essential to limit their occurrence and severity. The abuse and misuse of antibiotics [9], together with the prescription of unlicensed and off-label drugs [10,11], are among the most important causes of adverse event development, especially in children. In this case, the implementation of carefully planned stewardship programs can be effective in reducing the incidence of adverse events [9,12]. Antibiotic dosages are calculated on the basis of the pharmacokinetic and pharmacodynamic characteristics of each drug. Any modification of the absorption, distribution, metabolism and excretion of a drug due to disease, aging or organ immaturity may lead to a significant increase in the risk of antibiotic-related adverse events [13]. Antibiotic dosages that are well-tolerated and safe in healthy adult patients can be dangerous in sick subjects and in younger infants, as the amount of free drug that is able to exert an antimicrobial effect can significantly increase and reach toxic levels. The personalization of drug dosages according to the characteristics of the patient’s disease and their age and maturity can reduce the risk of adverse event development [13,14]. In order to achieve this goal, particularly for drugs that pose the highest risk, specific dosing tables are prepared to help determine the safe and effective dose for each condition and age.
In some subjects, such as children, antibiotics can interfere with tissue development and cause significant adverse events. The damage caused to both the cartilage in weight-bearing joints and the epiphyseal cartilage following the administration of fluoroquinolones [15], as well as the discoloration of permanent teeth following old tetracycline administration [16], are due to the increased sensitivity of the developing tissues to the antibiotic stimuli. These adverse events should be known, and the use of these drugs in at-risk subjects should be avoided whenever possible.
Finally, genetics can condition the development of antibiotic-related adverse events, and the screening of patients with supposed or demonstrated specific genetic mutations may reduce the incidence of drug-related adverse events. Recent studies have shown that the mutation of genes encoding drug-metabolizing enzymes and transporters, genetic variants of some components of the immune system or mutations of mitochondrial genes are potentially associated with significant modifications of drug disposition [17,18]. This can lead to variations in drug clearance, with reduced drug efficacy or accumulation and an increase in the risk of adverse events. Moreover, in some cases, toxic metabolites are formed. Finally, some genetic mutations are associated with an abnormal immune response, leading to specific tissue damage. For several drugs, the association between well-defined genetic mutations and an increase in the risk of adverse events has been definitively ascertained. This has led several institutions, including the Clinical Pharmacogenetics Implementation Consortium (CPIC), to publish genotype-based drug guidelines to help clinicians understand how available genetic test results could be used to optimize drug therapy in individual patients, according to the characteristics and frequency of genetic polymorphisms in the treated populations [19,20,21,22]. Moreover, health authorities have decided that the risk of genetically determined adverse events should be systematically included in the package leaflet of all the drugs for which this information is known. Regarding antibiotics, however, definitive conclusions have been drawn for very few molecules. For many antimicrobial drugs, the risk of genetically related adverse events has not been sufficiently demonstrated. In other cases, the relationship between the development of adverse events and specific mutations is well defined, but the risk is too low and the genetic screening process capable of identifying at-risk subjects is too complex to justify its introduction in clinical practice. Only for aminoglycosides are there sufficient data to suggest that pretherapy screening analysis should be performed; however, this is very difficult to implement.
2. Antibiotics for Which the Role of Genetics in Conditioning Development of Adverse Events Is Definitively Demonstrated
2.1. Aminoglycosides
Aminoglycosides (AGs) are an old class of antibiotics that include neomycin, streptomycin, gentamycin, netilmicin, tobramycin and amikacin among those most frequently prescribed. Figure 1 shows the chemical structure of AGs.
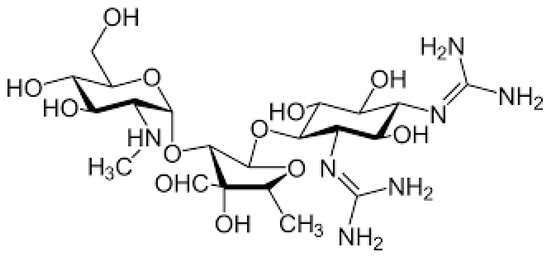
Figure 1. Chemical structure of aminoglycosides.
AGs exert concentration-dependent bactericidal activity against several Gram-negative aerobic pathogens and act synergistically with several other antibiotics against some clinically important Gram-positive bacteria, including Staphylococcus spp. [23]. The inhibition of bacterial protein synthesis is the main mechanism of action in AGs. They bind to the aminoacyl site of 16S ribosomal RNA within the 30S ribosomal subunit, promote the misreading of the bacterial genetic code and inhibit translocation. This results in error-prone protein synthesis that damages the membrane of the bacterial cell and leads to the death of the infectious agent [24]. Due to their large spectrum of activity, low cost and well-demonstrated persistent clinical efficacy, AGs continue to be frequently prescribed, alone or in combination with other antimicrobials, for the treatment of several suspected or documented life-threatening diseases in both immunocompetent and immunocompromised hosts of any age, including neonates [25]. Unfortunately, the use of AGs is not risk free, as it is frequently accompanied by the development of severe adverse events, among which nephrotoxicity and ototoxicity are the most important. Fortunately, renal damage is reversible; although it can lead to a transient increase in the serum concentration of AGs that favors the development of ototoxicity, it does not lead to permanent alterations in renal function, which returns to normal as soon as AG therapy is suspended. On the contrary, ototoxicity is associated with the development of permanent ear damage that involves the cochlea, the vestibule or both. Cochleotoxicity results in tinnitus and/or sensorineural hearing loss with permanent deafness. Vestibulotoxicity manifests as vertigo, nausea, nystagmus and ataxia. Streptomycin and gentamicin are mainly vestibulotoxic, while amikacin, neomycin and kanamycin are preferentially cochleotoxic. Tobramicin is equally vestibulotoxic and cochleotoxic [26,27].
To exert their cytotoxic effect, AGs must enter the tissue of the inner ear. When systematically administered, these drugs enter the endolymph from the bloodstream and are taken up by inner ear cells via mechanoelectrical transduction channels and apical endocytosis. In the ear cells, AGs bind to the 12S ribosomal RNA subunit of the mitochondrial ribosome and, due to the similarities between mammalian and bacterial ribosomes, interfere with human mitochondrial ribosomes in a similar manner to that in bacteria. When cell respiration is perturbed, the overproduction of superoxide occurs, together with cell apoptosis and the development of ear damage. Moreover, some AGs act on the composition of the otolithic membrane, thus changing its characteristics and causing vestibular damage [27]. Though the development of deafness represents a significant limitation regardless of the patient’s age, this clinical problem is considered a tragic event when it occurs in younger children as it can have a negative impact on language development, literacy, self-esteem and social skills [28]. A recent review of 29 studies carried out from 1975 to 2021, including seven randomized controlled trials, showed that up to 57% of children treated with AGs are at risk of the development of inner ear problems [29].
.2. Beta-Lactams
2.2.1. Amoxicillin–Clavulanic Acid
Amoxicillin–clavulanic acid (AC) is a combination of an antibiotic (amoxicillin) and a suicide inhibitor of bacterial beta-lactamases (clavulanic acid). The inhibition of these bacterial enzymes significantly extends the antibacterial activity of amoxicillin, making the combination effective against a large number of Gram-positive and Gram-negative infections. AC is indicated for the treatment of respiratory tract infections, urinary tract infections and skin and soft tissue infections. Moreover, the unapproved use of this combination in the treatment of several other supposed or documented bacterial infections is common worldwide [42]. In some countries, such as Italy, AC is the most common antibiotic regimen prescribed, especially in children [43]. AC is generally safe and well tolerated, with mild to moderate transient adverse events that are mainly associated with its effect on the gut microbiota. The only severe adverse event that has been reported is idiosyncratic drug-induced liver injury (DILI), which can develop 2 to 45 days after the initiation of therapy [44]. Studies have shown that DILI occurs in about 19.1 per 100,000 persons every year and that AC is the leading cause of this adverse event, with an incidence of 1.7 cases per every 10,000 prescriptions [45]. This frequency may be lower among the pediatric population; however, the data collected in children are very few and do not allow firm conclusions to be drawn in this regard [46]. The clavulanic acid component is considered the true cause of DILI, as the incidence of liver damage in patients receiving amoxicillin alone is significantly lower and not higher than approximately 0.3 cases per 10,000 prescriptions [47,48]. Liver damage can be hepatocellular, cholestatic or mixed, with hepatocellular injury predominating among children [45] and mixed injury predominating among older patients [47,48]. This damage is generally mild, as it regresses completely as soon as the drug is discontinued in most cases. In a very low number of patients, however, significant functional and structural liver alterations may develop, leading to the need for hospitalization and transplant and an increased risk of death [49]. It has been reported that about 17% of all the DILI cases leading to hospitalization are associated with the prescription of AC [49]. Regarding the pathogenesis of hepatotoxicity, it is thought that, in most cases, it depends on genetic variations that lead to an immunological reaction.
2.2.2. Flucloxacillin
Flucloxacillin (FC) is an isoxazolyl penicillin, a group of antibiotics that includes flucloxacillin, oxacillin, cloxacillin, dicloxacillin and methicillin [55]. Figure 2 shows the chemical structure of FC.
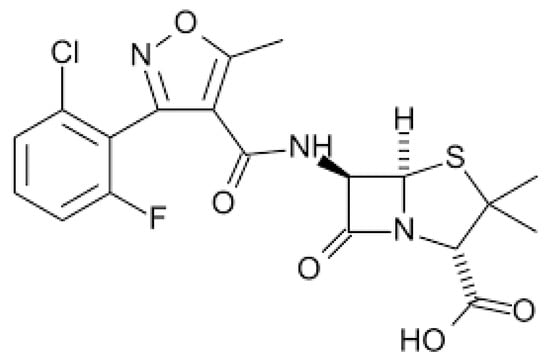
Figure 2. Chemical structure of flucloxacillin.
All these drugs are beta-lactamase resistant and have been largely used to treat infections due to Gram-positive rods, mainly penicillin-resistant Staphylococcus spp. Presently, due to the emergence of methicillin-resistant Staphylococcus strains, the use of flucloxacillin and other isoxazolyl penicillins has been significantly reduced [56].
FC is generally safe and well tolerated, as adverse events following its administration are low in frequency and are mild and transient. However, the administration of FC can lead to the development of cholestatic hepatitis, which occurs in 1–2 individuals per every 1,000 treated patients within 1 to 45 days of starting treatment [57]. This adverse event is relatively uncommon in children, as most cases have been diagnosed in patients older than 55 years [58]. In most cases, this liver disease tends to dissipate spontaneously in several months, even though the development of chronic vanishing bile duct syndrome is possible [59] and fatal cases have been described [60]. As for AC, immune pathogenesis is considered the basis for the development of liver disease caused by FC. It has been shown that more than 84% of patients with FC-associated hepatitis carry the HLA-B*57:01 allele, and that people with this genetic mutation have an 80 times greater likelihood of experiencing this adverse event [61,62,63]. Moreover, an association has also been seen with HLA-B*57:03 [62]. Interestingly, HLA-B*57 alleles have not been associated with hepatotoxicity induced by other isoxazolyl penicillins.
2.3. Antituberculous Drugs
Most patients, including children, suffering from tuberculosis are treated with a combination of multiple drugs; these are, most frequently, isoniazid (IS), rifampicin, pyrazinamide and ethambutol [65]. During treatment, generally between 6 weeks and 6 months after the start of drug administration [66], up to 20% of treated patients [67] develop signs of hepatotoxicity, with a lower frequency being observed among children [68,69]. Several factors, including age ≥60 years, female gender, a poor nutritional status and concomitant chronic hepatitis B infection, are associated with an increased risk of liver damage. However, genetic susceptibility seems to play a relevant role in this regard. In most cases, liver damage is limited to an asymptomatic, slight elevation in the concentration of serum transaminase in the liver that generally settles with the continued use of the drugs or disappears when the drugs are withdrawn. However, a subgroup accounting for approximately 1% of treated patients suffers from more severe drug-induced liver injury, with severe and prolonged transaminase elevation and relevant hepatocellular damage that can lead, in rare cases, to fulminant liver failure and death [70,71]. Although some cases of these adverse events have been ascribed to rifampicin [72] and pyrazinamide [73], IS is considered the most significant cause of liver damage in patients receiving antituberculous drugs [74]. Evidence suggesting that signs of hepatoxicity can be detected in patients of any age receiving isoniazid alone for prophylaxis strongly supports this conclusion [75].
2.4. Sulfonamides
Sulfonamides were the first synthetic antimicrobial drugs introduced in clinical practice [92]. They were originally active against several Gram-positive and Gram-negative bacteria, and were largely used against a large number of bacterial diseases with satisfactory results. They are bacteriostatic agents that act by competitively inhibiting folic acid synthesis, which prevents the growth and proliferation of bacteria [93]. With the availability of antibiotics that are bactericidal, more effective, better tolerated and safe, the prescription of sulfonamides progressively declined. Most of them are now rarely used in clinical practice. The only exception is sulfamethoxazole (SMX), which, in combination with trimethoprim (TMP-SMX), is indicated and largely prescribed for prophylaxis and the treatment of several bacterial infections, including traveler’s diarrhea, urinary tract infections, and shigellosis; it is also included by the World Health Organization (WHO) in the list of essential medicines [94].
3. Antibiotics for Which the Role of Genetics in Conditioning the Development of Adverse Events Is not Definitively Demonstrated
3.1. Linezolid
Linezolid (LZ) is the first oxazolidinone antibiotic to be produced; this is a group of drugs recently developed to overcome some of the clinical problems strictly related to the emergence of bacterial resistance to antibiotics. Figure 3 shows the chemical structure of LZ.
Figure 3. Chemical structure of linezolid.
LZ is effective against several Gram-positive drugs, including methicillin-resistant Staphylococcus aureus, and is indicated for the treatment of severe, life-threating conditions caused by these pathogens [102]. Moreover, as it is effective against multidrug-resistant or extensively-drug resistant Mycobacterium tuberculosis, LZ is included in the drug regimens used to treat patients infected with these resistant M. tuberculosis strains [103]. LZ exerts its antimicrobial activity via inhibition of bacterial protein synthesis in a way similar to that previously described for aminoglycosides. It binds to a site on the 23S ribosomal RNA of the 50S ribosomal subunit, thus preventing the formation of a functional 70S that is essential for the bacterial translation process [104]. Unfortunately, the long-term use of LZ is accompanied by the frequent development of several severe adverse events, including hyperlactatemia, lactic and metabolic acidosis, myelosuppression with thrombocytopenia and anemia, gastrointestinal disturbances and optic or peripheral neuropathy [105]. It is thought that, in most cases, these severe clinical problems depend, as in the case of AG, on the similarities between human and bacterial ribosomes. The inhibitory action exerted on protein synthesis and on bacterial ribosomes is extended to human ribosomes. On the other hand, in vitro studies have clearly shown that exposure to LZ significantly reduces the mitochondrial protein synthesis of mammalian cells [106,107]. Mitochondrial mutations may result in a predisposition to the development of LZ-related adverse events. Studies have shown that increased mitotoxicity and clinical symptoms can be found in patients harboring mtDNA haplogroup U, mutations in 12S rRNA or polymorphisms in the 16S rRNA sequence [108]. Moreover, polymorphisms in the ABCB1 or CYP3A genes have been associated with the significant modification of LZ clearance, which may play a role in conditioning the efficacy and tolerability of the drug [109]. However, no definitive conclusions in this regard have been drawn and no health authorities have so far made recommendations related to the genetic control of patients receiving this drug [110].
3.2. Fluoroquinolones
Fluoroquinolones (FQs) are a group of antibiotics with a broad spectrum of activity and excellent pharmacokinetics [111]. This explains why they were, initially, frequently used. In recent years, however, the prescription of FQs has been significantly reduced due to evidence suggesting that they could cause rare but very serious, disabling and potentially irreversible adverse events involving the musculoskeletal, nervous and psychiatric systems of the body. This led health authorities to restrict the use of these drugs only to very severe infections, unless other antibacterial medicines commonly recommended for these conditions could not be used [112,113]. Levofloxacin, ciprofloxacin, moxifloxacin, ofloxacin, gemifloxacin and delafloxacin are the FQs most frequently prescribed to treat severe and complicated urinary tract infections, intraabdominal infections, skin and soft tissue infections, community-acquired and nosocomial pneumonia and bone and joint infections [114]. Additionally, FQs such as moxifloxacin, gatifloxacin and levofloxacin are seeing increased off-label usage in the treatment of drug-resistant tuberculosis or cases of intolerance to other antituberculosis drugs [115]. The routine use of systemic FQs should be avoided in children due to the already reported potential risk of musculoskeletal toxicity. However, their off-label use, especially in children with cystic fibrosis or tuberculosis due to resistant bacteria, is relatively common [111].
It has been suggested that, at least in part, FQ-related adverse events are related to genetic variants that condition significant variations in drug disposition. FQs are substrates of the multiple ATP-binding cassette (ABC) superfamily of active transporters, which play a critical role in conditioning the passage of these drugs into tissues and across the blood–brain barrier [116,117]. In a case report, it was evidenced that a patient who had developed generalized seizures after treatment with levofloxacin carried polymorphisms of the efflux transporter genes ABCB1 and ABCG2, which code for P-glycoprotein and breast cancer resistance protein (BCRP), respectively. This could have conditioned the reduced activity of both these proteins and the increased passage of levofloxacin across the blood–brain barrier, causing seizures [118]. Polymorphisms of the ABCB1 gene and of the UDP glucuronosyltransferase family 1 member A1 (UGT1A1) gene were found in a small group of patients experiencing the reduced absorption of moxifloxacin, further supporting the hypothesis that genetics may condition the disposition of FQs and the development of unexpected adverse events [119]. This suspicion is further confirmed by the result of a study involving a second group of patients receiving moxifloxacin [120]. In this case, it was found that a significant increase in drug blood levels could be found in subjects carrying the -1187G>A variant in the solute carrier organic anion transporter family member 1B1 gene (SLCO1B1). This suggests that increased blood concentrations of the antibiotic could explain the prolongation of the QT interval and the other cardiac arrhythmias frequently reported among FQ-related adverse events. Although interesting, these findings do not definitively demonstrate a clear relationship between specific genetic variations and the development of some FQ-related adverse events. Further studies are needed to definitively clarify these problems.
3.3. Macrolides
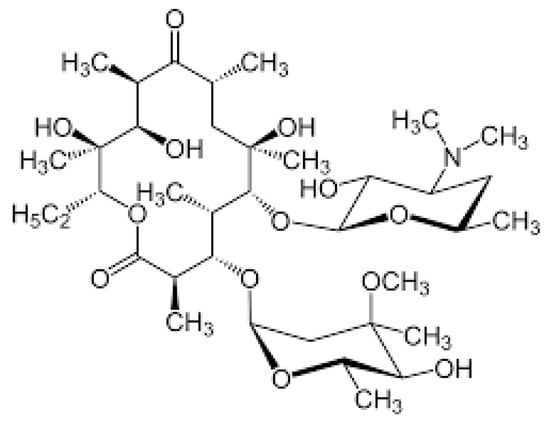
Macrolides (MCs) are a group of antibiotics characterized by a large lactone ring, which can vary from 12 to 16 atoms, with one or more sugar chains attached (Figure 4).
Figure 4. Chemical structure of macrolides.
The most used MCs are erythromycin (ER), clarithromycin (CL) and azithromycin (AZ). ER and CL are 14-membered macrolides, whereas AZ is a 16-membered drug [121]. Like several other antibiotics, MCs act via inhibition of bacterial protein synthesis. They interfere with ribosomal activity, binding to the bacterial 50S ribosomal subunit and preventing the translation of mRNA. Bacterial protein synthesis is consequently inhibited [122]. MCs are largely effective against several bacteria pathogens, including Streptococcus pyogenes, Streptococcus pneumoniae, Hemophilus influenzae, Bordetella pertussis and atypical bacteria. For this, they are considered an optimal solution for the treatment of respiratory infections and skin and soft tissue infections, particularly when penicillin cannot be used. Moreover, due to their activity against Helicobacter pylori, MCs have been introduced into a combination therapy for infections caused by this pathogen. Unfortunately, in recent years, a significant number of previously highly sensitive bacteria, especially S. pyogenes and S. pneumoniae, have developed resistance to MCs, and experts have recommended that these agents are prescribed only when the sensitivity of the infecting pathogen has not been previously established or is strongly suspected [123]. However, MCs have continued to be used in the treatment of atypical bacterial infection and Helicobacter pylori infections [124]. Generally, MCs are safe and well tolerated, although some relevant adverse events occasionally occur. The prolongation of the QT and QTc interval in the cardiac cycle, potentially favoring the development of cardiac arrhythmias like torsades de pointes, ventricular tachycardia, and ventricular fibrillation, is the most common adverse event reported [125].
This entry is adapted from the peer-reviewed paper 10.3390/ph17030331
This entry is offline, you can click here to edit this entry!