Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Sudden cardiac death (SCD) accounts for a substantial proportion of mortality in heart failure with reduced ejection fraction (HFrEF), frequently triggered by ventricular arrhythmias (VA). Guideline-directed medical therapy (GDMT) for HFrEF is founded on a cornerstone of four distinct classes of medications: beta-blockers (BB), renin-angiotensin-aldosterone system (RAAS) inhibitors, mineralocorticoid receptor antagonists (MRA), and sodium-glucose cotransporter 2 inhibitors (SGLT2i).
- heart failure with reduced ejection fraction
- sudden cardiac death
- ventricular arrhythmias
- guideline-directed medical therapy
- pharmacological management
1. Introduction
Heart failure (HF), and, more specifically, heart failure with reduced ejection fraction (HFrEF), stands as a public health challenge characterized by substantial morbidity and mortality, notably attributed to sudden cardiac death (SCD). The incidence of SCD in the HF population is fivefold greater than in the general population, accounting for 40–50% of all deaths, which is a considerable proportion of potentially preventable deaths in this population [1]. The association between SCD and HF is predominantly mediated by ventricular arrhythmias (VA), resulting from the complex interplay between the structurally altered cardiac substrate in HF and various environmental triggers [2][3].
Guideline-directed medical therapy (GDMT) for HFrEF is founded on a cornerstone of four distinct classes of medications: beta-blockers (BB), renin-angiotensin-aldosterone system (RAAS) inhibitors, mineralocorticoid receptor antagonists (MRA), and sodium-glucose cotransporter 2 inhibitors (SGLT2i) [4]. Each of these pharmacological classes has demonstrated significant efficacy in reducing major cardiac events in HFrEF patients, including, for some, reduction in the incidence of SCD [5][6].
2. Pathophysiology of Ventricular Arrhythmia and Sudden Cardiac Death
The pathophysiology of SCD due to VA in the context of HF is multifaceted. It involves an intricate interplay among transient factors or events that serve as triggers (e.g., ischemic episodes, metabolic disturbances, electrolyte imbalances, fluctuations in sympathetic nervous system activity), occurring in the presence of a myocardial substrate predisposed to arrhythmias [3][7].
Three primary electrophysiological mechanisms lead to VA within this context: (1) Inappropriately Increased Automaticity: This mechanism involves certain ventricular myocyte regions exhibiting increased automaticity, thereby initiating spontaneous action potentials without external stimulation, contributing to the onset of arrhythmias. (2) Triggered Activity: Triggered activity can manifest as early afterdepolarizations (occurring late in phase 2 or early in phase 3 of the action potential) or delayed afterdepolarizations (occurring after complete repolarization). These afterdepolarizations can reach the threshold potential necessary for the activation of ion channels, ultimately leading to arrhythmic events. Triggered activity typically follows a preceding impulse and is not a self-generating rhythm. (3) Reentry: This involves the circulation of electrical impulses around a barrier, often anatomical, such as scar tissue or fibrosis, perpetuating a continuous myocardial excitation, leading to sustained arrhythmias. It is noteworthy that these three mechanisms can coexist; arrhythmias may initiate through triggered activity and transition to reentrant patterns [7][8][9].
The pathophysiological changes in HF create an environment that favors these VA mechanisms. HF is characterized by cardiac remodeling, which includes ventricular enlargement, hypertrophy, and fibrosis [10]. Fibrosis may result from myocardial infarctions or from other structural cardiac disorders such as hypertrophic cardiomyopathy and arrhythmogenic cardiomyopathy. Factors like TGF-β1, endothelin-1, and Angiotensin II induce fibrosis, perturbing electrical excitation and repolarization [11]. Regions of fibrosis facilitate reentrant tachycardias by disrupting myocardial electrical impulses and fostering areas of slow conduction [12].
The nervous system and the RAAS, which are both markedly activated in the setting of HFrEF due to ventricular dysfunction, are significantly implicated in arrhythmogenesis. Angiotensin II and Aldosterone, acting through RAAS, induce vasoconstriction, increasing the afterload and remodeling, and stimulate fibroblasts’ activity, leading to interstitial fibrosis and scar formation [10][13]. At the cellular level, angiotensin II initiates multiple signaling pathways that induce electrical remodeling, notably through alterations in the sodium current [14] and the destabilization of Kv4.3 messenger RNA [15], leading to increased Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity, perturbing calcium homeostasis, and predisposing to VA [16][17][18]. Moreover, Angiotensin II stimulates the sympathetic nervous system, contributing to VA through an increased sympathetic tone. Elevated levels of norepinephrine raise the afterload, thus increasing the myocardial oxygen demand and promoting cardiac remodeling, dilatation, and fibrosis [10]. Such structural alterations create a favorable environment for arrhythmogenesis. Additionally, sympathetic activation induces electrophysiological alterations, disrupting sodium and calcium channel currents and prolonging action’s potential duration, ultimately predisposing the myocardium to early depolarizations [19]. For example, chronic adrenergic activation destabilizes Ryanodine receptors 2 (RyR2) channels, which are critical Ca2+ release channels located on the sarcoplasmic reticulum, playing a central role in the excitation-contraction coupling in the heart. Dysfunctional RyR2 channels lead to calcium leak and elevated cytoplasmic calcium levels via CaMKII, impairing cardiac function and exacerbating arrhythmogenesis through electrical instability, delayed afterdepolarizations, and triggered arrhythmias [18][19].
All this intricate electrical and structural remodeling in HFrEF leads to arrhythmogenesis and SCD (Figure 1). This highlights the importance of the GDMT for HFrEF in mitigating the risk of SCD in this population.
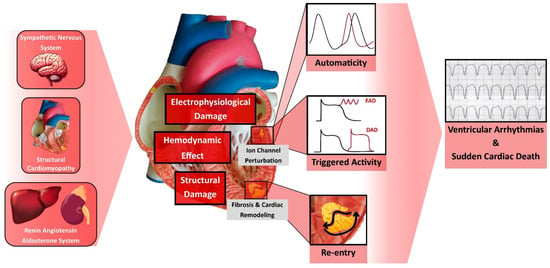
Figure 1. Pathophysiology of Ventricular Arrhythmia and Sudden Cardiac Death in Heart Failure. DAD, Delayed Afterdepolarization; EAD, Early Afterdepolarization.
3. Beta-Blockers
β-adrenergic receptors are expressed on cardiomyocytes. The activation of the cardiac sympathetic system by the binding of norepinephrine and epinephrine to these receptors initiates a cascade of intracellular reactions involving the cAMP/PKA signaling pathway [20]. This signaling pathway affects several key targets, including RyR2, L-type calcium channels, phospholamban, and others [19][20]. The net result of these signaling events is the enhancement of various aspects of cardiac function: an increased heart rate (chronotropy), an accelerated conduction velocity (dromotropy), a heightened force of contraction (inotropy), and an improved speed of relaxation (lusitropy) [21]. All these downstream effects of β-adrenergic stimulation facilitate the development of ectopic activity, including early afterdepolarizations and delayed afterdepolarizations, as well as functional reentry through action potential duration shortening, effective refractory period reduction, and conduction alterations [21]. Consequently, excessive β-adrenergic stimulation is associated with arrhythmias.
Three subtypes of β-receptors are known, with β1-receptors being predominantly localized in the cardiac tissue. The classes of BB are diverse, encompassing non-selective β-adrenergic antagonists (e.g., nadolol, propranolol), β1-selective adrenergic antagonists (e.g., acebutolol, atenolol, esmolol, metoprolol), and β-adrenergic antagonists with additional cardiovascular effects (e.g., carvedilol, labetalol, nebivolol), which may exhibit vasodilatory, anti-inflammatory, and antioxidant properties [22]. BB attenuate the sympathetic tone and sympathetically mediated triggers [22]. They mitigate automaticity by prolonging the sinus node cycle length and decelerating the atrioventricular conduction velocity by limiting calcium entry via catecholamine-dependent channels [21]. Moreover, BB have been shown to possess antifibrotic properties, improve the left ventricular (LV) function, reduce the LV diameter, and decrease the LV mass, outcomes that are favorable in reducing SCD in HF [21].
Multiple studies have demonstrated the efficacy of BB in preventing arrhythmias and SCD in the context of HF. CIBIS II investigated the impact of Bisoprolol in HFrEF and demonstrated a significant mortality benefit, with a 42% reduction in SCD compared to the placebo group [23]. Metoprolol in HFrEF showed a similar and significant reduction in SCD in the MERIT-HF trial [24]. In the SENIORS trial, evaluating Nebivolol in an elderly HFrEF population, the SCD rate was significantly lower in the nebivolol-treated group [25]. The COPERNICUS trial in 2001, studying Carvedilol, showed a mortality benefit, although explicit data on SCD were not presented [26]. Additionally, several studies have indicated a significant reduction in shocks in patients wearing implantable cardioverter–defibrillators (ICD) when treated with BB compared to those without [27]. Meta-analyses have confirmed the benefit of BB on both mortality and SCD, without revealing statistically significant differences among the various types of BB [28][29][30]. Recent evidence, however, indicates a potential superiority of non-selective beta-blockers in antiarrhythmic effects. Specifically, Propranolol has been demonstrated to be superior to Metoprolol in managing electrical storms in ICD patients [31]. Carvedilol has also been associated with a substantial decrease in VA risk, compared to Metoprolol [32]. Further robust data are needed to confirm these findings.
In conclusion, BB substantially reduce proarrhythmic risk by inhibiting sympathetically mediated triggers, reducing functional reentrant substrates, and slowing the sinus node and atrioventricular nodal rates. The evidence supporting their use in HFrEF is robust and emphasizes their critical role in mitigating arrhythmias and SCD in this patient population.
4. Angiotensin-Converting Enzyme Inhibitor/Angiotensin Receptor Blocker
The RAAS exerts a critical influence on the pathophysiology of HFrEF by contributing to cardiac remodeling, interstitial fibrosis, and perturbations in various ionic currents, which collectively enhance arrhythmogenesis. The inhibition of the RAAS is principally achieved through two pharmacological classes: angiotensin-converting enzyme inhibitors (ACEi) and angiotensin II receptor blockers (ARB). ACEi act by inhibiting the conversion of angiotensin I to angiotensin II, while ARB selectively antagonize the angiotensin II type 1 receptors [33]. This blockage results in several beneficial effects, including the attenuation of ventricular remodeling, vasodilatation, a reduction in neurohormonal agents, and a decrease in the sympathetic tone and circulating catecholamines, along with favorable effects on ionic currents [10][34].
Several pivotal trials have explored the efficacy of RAAS inhibitors in the context of HFrEF. The CONSENSUS study, assessing the use of Enalapril in HFrEF, demonstrated a significant reduction in mortality, although it did not find a difference in SCD [35]. Similar results were found in the SOLVD-Prevention study [36]. In the ELITE trial, which compared Losartan to Captopril, Losartan was associated with a lower mortality, primarily driven by a lower incidence of SCD [37]. However, these results were not replicated in the ELITE II trial, where no significant differences were observed in either mortality or SCD [38]. Trials such as CHARM (exploring Candesartan) [39] and Val-HEFT (exploring Valsartan) [40] did not specifically monitor SCD as an endpoint. In patients wearing ICD, ACEi/ARB therapy has been associated with an improved freedom from shocks [41][42]. A large retrospective registry by Schupp et al. [43] indicated that ACEI/ARB treatment is associated with reduced all-cause mortality in patients who survived episodes of VA. Nonetheless, meta-analyses have shown a lack of a consistent efficacy of ACEi/ARBs in reducing the risk of SCD and VA, despite demonstrating benefits in overall mortality and hospitalization rates [30][44][45][46]. The exception appears to be in the context following acute myocardial infarction, where ACEi have shown a 20% reduction in SCD [47].
In conclusion, although ACEi and ARB have demonstrated favorable effects on the RAAS and have robust data supporting their beneficial impact on overall outcomes in HFrEF, their role in reducing the risk of SCD has not been definitively established, as underscored by multiple meta-analyses.
5. Angiotensin Receptor–Neprilysin Inhibitor
The Angiotensin Receptor–Neprilysin Inhibitor (ARNi) combines two active components: Valsartan, an ARB, and Sacubitril, a Neprilysin Inhibitor [48]. The molecular mechanisms underlying the anti-arrhythmic effects of ARNi are not yet fully elucidated and remain speculative to some extent. The mechanism of action encompasses effects on the RAAS via the ARB component and additional effects attributable to the Neprilysin Inhibitor, Sacubitril. At the electrophysiological level, ARNi exerts an influence on ionic currents in cardiomyocytes. Chang et al. [49] observed that ARNi had the potential to up-regulate the expression of potassium channel proteins, including KCNH2, KCNE1, and KCNE2. The consequent shortening of the action potential duration potentially ameliorates ventricular arrhythmogenicity, especially in the context of HF induced by myocardial infarction. Moreover, ARNi also appears to have a favorable effect on calcium homeostasis by downregulating the expression of CaMKII and mitigating diastolic calcium leak arising from dysfunctional RyR2 [50][51]. Additionally, neprilysin inhibition by ARNi contributes to elevated circulating levels of natriuretic peptides, which exert various cardioprotective effects countering the detrimental effects of the RAAS and sympathetic nervous system activation [52]. Natriuretic peptides increase the intracellular levels of cyclic guanosine monophosphate and its downstream effector molecule protein kinase G. This cascade leads to vasodilation, natriuresis, the inhibition of the RAAS, and sympathetic systems [52]. Furthermore, natriuretic peptides exert anti-inflammatory, anti-apoptotic, anti-hypertrophic, and anti-fibrotic effects on the myocardium [53][54][55]. ARNi has also been shown to significantly reduce biomarkers associated with profibrotic signaling [56].
Multiple studies and trials corroborate the beneficial effects of ARNi on VA and ICD therapy [57][58]. Post hoc analyses of the PARADIGM-HF trial, investigating ARNi in HFrEF, showed a significant reduction in VA and the risk of SCD [59][60]. Liu et al. [61] found in their meta-analyses that although ARNi did not affect the incidence of VA, it did significantly reduce the risk of SCD in heart failure patients. Another meta-analysis by Pozzi et al. [62] demonstrated a significant reduction in the burden of VA and ICD shock when comparing ARNi to ACEi/ARB therapy. Fernandes et al. [63] reported a significant reduction in SCD, VA, and appropriate ICD therapy with ARNi. Major studies and meta-analyses regarding the effect of ARNi on VA and SCD are summarized in Table 1.
Table 1. Major studies and meta-analyses regarding effect of ARNi on ventricular arrythmia and sudden cardiac death.
| Authors Journal Year |
Type of Study Intervention |
No. of Patients in the Population |
Effect on VA and SCD |
|---|---|---|---|
| Martens, et al. Clin Res Cardiol. 2019 [57]. |
Retrospective, cohort Pre- vs. Post-ARNi initiation |
151 HFrEF with ICD |
VA reduction (51 vs. 14; p < 0.001) ICD shock reduction (16 vs. 6; p < 0.001) |
| De Diego, et al. Heart Rhythm. 2018 [58]. |
Prospective, cohort ARNi vs. ACEi/ARB |
240 HFrEF with ICD |
VA and ICD shock reduction (0.8% vs. 6.7%; p < 0.02) |
| Russo, et al. J Clin Med. 2020 [64]. |
Prospective, cohort ARNi |
167 HFrEF with ICD |
VA reduction (15 vs. 4; p = 0.03) ICD shock reduction (13 vs. 3; p = 0.02) |
| Rohde et al. JACC Heart Fail. 2020 [59]. |
RCT—post hoc analysis ARNi vs. ACEi |
8399 HFrEF |
SCD reduction in the ICD group (HR 0.49; 95% CI 0.25–0.99) and non-ICD group (HR 0.81; 95% CI 0.67–0.98) |
| Curtain, et al. Eur J Heart Fail. 2022 [60]. |
RCT—post hoc analysis ARNi vs. ACEi |
8399 HFrEF |
VA reduction (HR 0.76; 95% CI 0.62–0.95) |
| Fernandes, et al. Heart Rhythm O2. 2021 [63]. |
Meta-analysis ARNi vs. ACEi/ARB |
11,204 HFrEF |
SCD reduction (OR 0.78; 95% CI 0.63–0.96) VA reduction (OR 0.45; 95% CI 0.25–0.79) Higher BiV Pacing (p < 0.0001) |
| Liu, et al. Front Cardiovasc Med. 2022 [61]. |
Meta-analysis ARNi vs. ACEi/ARB/Placebo |
18,500 HFrEF or HFpEF |
No VA reduction (RR 0.86; 95% CI 0.68–1.10) SCD reduction (RR 0.79; 95% CI 0.70–0.90) |
| Mujadzic, et al. J Innoc Card Rhythm Mang. 2022 [65]. |
Meta-analysis ARNi vs. ACEi/ARB/Placebo |
18,548 HFrEF or HFpEF |
VA & SCD reduction (OR 0.71; 95% CI 0.54–0.93) ICD shock reduction (OR 0.23; 95% CI 0.11–0.47) |
| Pozzi, et al. Heart Fail Rev. 2023 [62]. |
Meta-analysis ARNi vs. ACEi/ARB |
8837 HFrEF |
VA reduction (OR 0.78; 95% CI 0.63–0.96 for RCT and RR 0.62; 95% CI 0.53–0.72 for observational studies) ICD shock reduction (RR 0.24; 95% CI 0.12–0.24) |
ACEi, Angiotensin-Converting Enzyme Inhibitor; ARB, Angiotensin II Receptor Blocker; ARNi, Angiotensin Receptor–Neprilysin Inhibitor; BiV, Biventricular; HFpEF, Heart Failure with Preserved Ejection Fraction; HFrEF, Heart Failure with Reduced Ejection Fraction; ICD, Implantable Cardioverter–Defibrillator; RCT, Randomized Controlled Trial; SCD, Sudden Cardiac Death; VA, Ventricular Arrhythmia.
In conclusion, ARNi therapy appears to manifest favorable effects via multiple mechanisms, including vasodilation, the attenuation of sympathetic activation, the reduction in myocardial wall stretch and fibrosis, and modulatory impacts on ion channels such as potassium channels, RyR2, and the CaMKII pathway. Meta-analyses confirm ARNi’s efficacy in reducing both VA and SCD, yet further investigation is needed to fully understand the precise molecular mechanisms.
6. Mineralocorticoid Receptor Antagonists
MRA provide a complementary approach to the neurohormonal suppression of the RAAS by targeting the aldosterone receptor [66]. These agents achieve a series of beneficial cardiovascular outcomes, including the prevention of the electrical remodeling of cardiac tissue [67][68], the attenuation of myocardial fibrosis and ventricular remodeling [69][70], a decrease in sympathetic activation [71], and beneficial effects on endothelial vasomotor dysfunction [72]. Through these several mechanisms, MRA have been shown to prevent SCD.
The RALES trial, which investigated the use of Spironolactone in patients with HFrEF, reported a significant reduction in both overall mortality and cardiac-specific mortality. It showed a 29% reduction in the risk of SCD [73]. Similarly, the EPHESUS trial focusing on Eplerenone demonstrated a significant reduction in all-cause mortality, cardiovascular-related death, and the risk of cardiovascular-related death or hospitalization. Notably, it also showed a significant reduction in the incidence of SCD [74]. Numerous meta-analyses have consistently shown a clear benefit for MRA in reducing SCD, reinforcing their critical role in managing patients with HFrEF [30][75][76][77].
This entry is adapted from the peer-reviewed paper 10.3390/jcm13051316
References
- Kannel, W.B.; Plehn, J.F.; Cupples, L. Cardiac failure and sudden death in the Framingham Study. Am. Heart J. 1988, 115, 869–875.
- Zipes, D.P.; Wellens, H.J.J. Sudden Cardiac Death. Circulation 1998, 98, 2334–2351.
- Deo, R.; Albert, C.M. Epidemiology and Genetics of Sudden Cardiac Death. Circulation 2012, 125, 620–637.
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. Heart J. 2021, 42, 3599–3726.
- Tromp, J.; Ouwerkerk, W.; van Veldhuisen, D.J.; Hillege, H.L.; Richards, A.M.; van der Meer, P.; Anand, I.S.; Lam, C.S.; Voors, A.A. A Systematic Review and Network Meta-Analysis of Pharmacological Treatment of Heart Failure with Reduced Ejection Fraction. JACC Heart Fail. 2022, 10, 73–84.
- Shen, L.; Jhund, P.S.; Petrie, M.C.; Claggett, B.L.; Barlera, S.; Cleland, J.G.F.; Dargie, H.J.; Granger, C.B.; Kjekshus, J.; Køber, L.; et al. Declining Risk of Sudden Death in Heart Failure. N. Engl. J. Med. 2017, 377, 41–51.
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126.
- Cherry, E.M.; Fenton, F.H.; Gilmour, R.F. Mechanisms of ventricular arrhythmias: A dynamical systems-based perspective. Am. J. Physiol. Circ. Physiol. 2012, 302, H2451–H2463.
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993.
- Schrier, R.W.; Abraham, W.T. Hormones and Hemodynamics in Heart Failure. N. Engl. J. Med. 1999, 341, 577–585.
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149.
- Verheule, S.; Schotten, U. Electrophysiological Consequences of Cardiac Fibrosis. Cells 2021, 10, 3220.
- Weber, K.T.; Brilla, C.G. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 1991, 83, 1849–1865.
- Mathieu, S.; El Khoury, N.; Rivard, K.; Gélinas, R.; Goyette, P.; Paradis, P.; Nemer, M.; Fiset, C. Reduction in Na+ current by angiotensin II is mediated by PKCα in mouse and human-induced pluripotent stem cell–derived cardiomyocytes. Heart Rhythm. 2016, 13, 1346–1354.
- Zhang, T.-T.; Takimoto, K.; Stewart, A.F.R.; Zhu, C.; Levitan, E.S. Independent Regulation of Cardiac Kv4.3 Potassium Channel Expression by Angiotensin II and Phenylephrine. Circ. Res. 2001, 88, 476–482.
- Zhao, Z.; Fefelova, N.; Shanmugam, M.; Bishara, P.; Babu, G.J.; Xie, L.-H. Angiotensin II induces afterdepolarizations via reactive oxygen species and calmodulin kinase II signaling. J. Mol. Cell. Cardiol. 2011, 50, 128–136.
- Sag, C.M.; Wadsack, D.P.; Khabbazzadeh, S.; Abesser, M.; Grefe, C.; Neumann, K.; Opiela, M.-K.; Backs, J.; Olson, E.N.; Brown, J.H.; et al. Calcium/Calmodulin-Dependent Protein Kinase II Contributes to Cardiac Arrhythmogenesis in Heart Failure. Circ. Heart Fail. 2009, 2, 664–675.
- Shannon, T.R.; Lew, W.Y. Diastolic Release of Calcium from the Sarcoplasmic Reticulum: A Potential Target for Treating Triggered Arrhythmias and Heart Failure. J. Am. Coll. Cardiol. 2009, 53, 2006–2008.
- Bers, D.M. Calcium Cycling and Signaling in Cardiac Myocytes. Annu. Rev. Physiol. 2008, 70, 23–49.
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205.
- Grandi, E.; Ripplinger, C.M. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol. Res. 2019, 146, 104274.
- Barrese, V.; Taglialatela, M. New advances in beta-blocker therapy in heart failure. Front. Physiol. 2013, 4, 323.
- CIBIS-II Investigators and committees The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): A randomised trial. Lancet 1999, 353, 9–13.
- Merit-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in-Congestive Heart Failure (MERIT-HF). Lancet 1999, 353, 2001–2007.
- Flather, M.D.; Shibata, M.C.; Coats, A.J.; Van Veldhuisen, D.J.; Parkhomenko, A.; Borbola, J.; Cohen-Solal, A.; Dumitrascu, D.; Ferrari, R.; Lechat, P.; et al. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur. Heart J. 2005, 26, 215–225.
- Packer, M.; Coats, A.J.; Fowler, M.B.; Katus, H.A.; Krum, H.; Mohacsi, P.; Rouleau, J.L.; Tendera, M.; Castaigne, A.; Roecker, E.B.; et al. Effect of Carvedilol on Survival in Severe Chronic Heart Failure. N. Engl. J. Med. 2001, 344, 1651–1658.
- Connolly, S.J.; Dorian, P.; Roberts, R.S.; Gent, M.; Bailin, S.; Fain, E.S.; Thorpe, K.; Champagne, J.; Talajic, M.; Coutu, B.; et al. Comparison of β-Blockers, Amiodarone Plus β-Blockers, or Sotalol for Prevention of Shocks from Implantable Cardioverter Defibrillators: The OPTIC Study: A Randomized Trial. JAMA 2006, 295, 165–171.
- Chatterjee, S.; Biondi-Zoccai, G.; Abbate, A.; D’ascenzo, F.; Castagno, D.; Van Tassell, B.; Mukherjee, D.; Lichstein, E. Benefits of blockers in patients with heart failure and reduced ejection fraction: Network meta-analysis. BMJ 2013, 346, f55.
- Al-Gobari, M.; El Khatib, C.; Pillon, F.; Gueyffier, F. Beta-blockers for the prevention of sudden cardiac death in heart failure patients: A meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2013, 13, 52.
- Al-Gobari, M.; Al-Aqeel, S.; Gueyffier, F.; Burnand, B. Effectiveness of drug interventions to prevent sudden cardiac death in patients with heart failure and reduced ejection fraction: An overview of systematic reviews. BMJ Open 2018, 8, e021108.
- Chatzidou, S.; Kontogiannis, C.; Tsilimigras, D.I.; Georgiopoulos, G.; Kosmopoulos, M.; Papadopoulou, E.; Vasilopoulos, G.; Rokas, S. Propranolol Versus Metoprolol for Treatment of Electrical Storm in Patients with Implantable Cardioverter-Defibrillator. J. Am. Coll. Cardiol. 2018, 71, 1897–1906.
- Diamond, A.; Goldenberg, I.; Younis, A.; Goldenberg, I.; Sampath, R.; Kutyifa, V.; Chen, A.Y.; McNitt, S.; Polonsky, B.; Steinberg, J.S.; et al. Effect of Carvedilol vs Metoprolol on Atrial and Ventricular Arrhythmias Among Implantable Cardioverter-Defibrillator Recipients. JACC Clin. Electrophysiol. 2023, 9, 2122–2131.
- Goodfriend, T.L.; Elliott, M.E.; Catt, K.J. Angiotensin Receptors and Their Antagonists. N. Engl. J. Med. 1996, 334, 1649–1655.
- Makkar, K.M.; Sanoski, C.A.; Spinler, S.A. Role of Angiotensin-Converting Enzyme Inhibitors, Angiotensin II Receptor Blockers, and Aldosterone Antagonists in the Prevention of Atrial and Ventricular Arrhythmias. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2009, 29, 31–48.
- CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med. 1987, 316, 1429–1435.
- SOLVD Investigators; Yusuf, S.; Pitt, B.; Davis, C.E.; Hood, W.B.; Cohn, J.N. Effect of Enalapril on Survival in Patients with Reduced Left Ventricular Ejection Fractions and Congestive Heart Failure. N. Engl. J. Med. 1991, 325, 293–302.
- Pitt, B.; Segal, R.; Martinez, F.A.; Meurers, G.; Cowley, A.J.; Thomas, I.; Deedwania, P.C.; Ney, D.; Snavely, D.B.; Chang, P.I. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE). Lancet 1997, 349, 747–752.
- Pitt, B.; Poole-Wilson, P.A.; Segal, R.; Martinez, F.A.; Dickstein, K.; Camm, A.J.; Konstam, M.A.; Riegger, G.; Klinger, G.H.; Neaton, J.; et al. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure: Randomised trial—The Losartan Heart Failure Survival Study ELITE II. Lancet 2000, 355, 1582–1587.
- McMurray, J.J.; Östergren, J.; Swedberg, K.; Granger, C.B.; Held, P.; Michelson, E.L.; Olofsson, B.; Yusuf, S.; Pfeffer, M.A. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: The CHARM-Added trial. Lancet 2003, 362, 767–771.
- Cohn, J.N.; Tognoni, G. Valsartan Heart Failure Trial Investigators. A Randomized Trial of the Angiotensin-Receptor Blocker Valsartan in Chronic Heart Failure. N. Engl. J. Med. 2001, 345, 1667–1675.
- AlJaroudi, W.A.; Refaat, M.M.; Habib, R.H.; Al-Shaar, L.; Singh, M.; Gutmann, R.; Bloom, H.L.; Dudley, S.C.; Ellinor, P.T.; Saba, S.F.; et al. Effect of Angiotensin-Converting Enzyme Inhibitors and Receptor Blockers on Appropriate Implantable Cardiac Defibrillator Shock in Patients with Severe Systolic Heart Failure (from the GRADE Multicenter Study). Am. J. Cardiol. 2015, 115, 924–931.
- Schupp, T.; Behnes, M.; Weiß, C.; Nienaber, C.; Lang, S.; Reiser, L.; Bollow, A.; Taton, G.; Reichelt, T.; Ellguth, D.; et al. Prognostic Impact of Angiotensin-Converting Enzyme Inhibitors and Receptor Blockers on Recurrent Ventricular Tachyarrhythmias and Implantable Cardioverter–Defibrillator Therapies. J. Cardiovasc. Pharmacol. 2019, 73, 272–281.
- Schupp, T.; Behnes, M.; Weiß, C.; Nienaber, C.; Lang, S.; Reiser, L.; Bollow, A.; Taton, G.; Reichelt, T.; Ellguth, D.; et al. Beta-Blockers and ACE Inhibitors Are Associated with Improved Survival Secondary to Ventricular Tachyarrhythmia. Cardiovasc. Drugs Ther. 2018, 32, 353–363.
- Garg, R.; Yusuf, S.; Bussmann, W.D.; Sleight, P.; Uprichard, A.; Massie, B.; McGrath, B.; Nilsson, B.; Pitt, B.; Magnani, B.; et al. Overview of Randomized Trials of Angiotensin-Converting Enzyme Inhibitors on Mortality and Morbidity in Patients with Heart Failure. Collaborative Group on ACE Inhibitor Trials. JAMA 1995, 273, 1450–1456.
- Flather, M.D.; Yusuf, S.; Køber, L.; Pfeffer, M.; Hall, A.; Murray, G.; Torp-Pedersen, C.; Ball, S.; Pogue, J.; Moyé, L.; et al. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: A systematic overview of data from individual patients. ACE-Inhibitor Myocardial Infarction Collaborative Group. Lancet 2000, 355, 1575–1581.
- Tsartsalis, D.; Korela, D.; Karlsson, L.O.; Foukarakis, E.; Svensson, A.; Anastasakis, A.; Venetsanos, D.; Aggeli, C.; Tsioufis, C.; Braunschweig, F.; et al. Risk and Protective Factors for Sudden Cardiac Death: An Umbrella Review of Meta-Analyses. Front. Cardiovasc. Med. 2022, 9, 848021.
- Domanski, M.J.; Exner, D.V.; Borkowf, C.B.; Geller, N.L.; Rosenberg, Y.; Pfeffer, M.A. Effect of angiotensin converting enzyme inhibition on sudden cardiac death in patients following acute myocardial infarction: A meta-analysis of randomized clinical trials. J. Am. Coll. Cardiol. 1999, 33, 598–604.
- Gu, J.; Noe, A.; Chandra, P.; Al-Fayoumi, S.; Ligueros-Saylan, M.; Sarangapani, R.; Maahs, S.; Ksander, G.; Rigel, D.F.; Jeng, A.Y.; et al. Pharmacokinetics and Pharmacodynamics of LCZ696, a Novel Dual-Acting Angiotensin Receptor—Neprilysin Inhibitor (ARNi). J. Clin. Pharmacol. 2010, 50, 401–414.
- Chang, P.-C.; Lin, S.-F.; Chu, Y.; Wo, H.-T.; Lee, H.-L.; Huang, Y.-C.; Wen, M.-S.; Chou, C.-C. LCZ696 Therapy Reduces Ventricular Tachyarrhythmia Inducibility in a Myocardial Infarction-Induced Heart Failure Rat Model. Cardiovasc. Ther. 2019, 2019, 6032631.
- Eiringhaus, J.; Wünsche, C.M.; Tirilomis, P.; Herting, J.; Bork, N.; Nikolaev, V.O.; Hasenfuss, G.; Sossalla, S.; Fischer, T.H. Sacubitrilat reduces pro-arrhythmogenic sarcoplasmic reticulum Ca2+ leak in human ventricular cardiomyocytes of patients with end-stage heart failure. ESC Heart Fail. 2020, 7, 2992–3002.
- Chang, P.-C.; Wo, H.-T.; Lee, H.-L.; Lin, S.-F.; Chu, Y.; Wen, M.-S.; Chou, C.-C. Sacubitril/Valsartan Therapy Ameliorates Ventricular Tachyarrhythmia Inducibility in a Rabbit Myocardial Infarction Model. J. Card. Fail. 2020, 26, 527–537.
- D’Elia, E.; Iacovoni, A.; Vaduganathan, M.; Lorini, F.L.; Perlini, S.; Senni, M. Neprilysin inhibition in heart failure: Mechanisms and substrates beyond modulating natriuretic peptides. Eur. J. Heart Fail. 2017, 19, 710–717.
- Pfau, D.; Thorn, S.L.; Zhang, J.; Mikush, N.; Renaud, J.M.; Klein, R.; deKemp, R.A.; Wu, X.; Hu, X.; Sinusas, A.J.; et al. Angiotensin Receptor Neprilysin Inhibitor Attenuates Myocardial Remodeling and Improves Infarct Perfusion in Experimental Heart Failure. Sci. Rep. 2019, 9, 5791.
- von Lueder, T.G.; Wang, B.H.; Kompa, A.R.; Huang, L.; Webb, R.; Jordaan, P.; Atar, D.; Krum, H. Angiotensin Receptor Neprilysin Inhibitor LCZ696 Attenuates Cardiac Remodeling and Dysfunction After Myocardial Infarction by Reducing Cardiac Fibrosis and Hypertrophy. Circ. Heart Fail. 2015, 8, 71–78.
- Sung, Y.-L.; Lin, T.-T.; Syu, J.-Y.; Hsu, H.-J.; Lin, K.-Y.; Liu, Y.-B.; Lin, S.-F. Reverse electromechanical modelling of diastolic dysfunction in spontaneous hypertensive rat after sacubitril/valsartan therapy. ESC Heart Fail. 2020, 7, 4040–4050.
- Zile, M.R.; O’Meara, E.; Claggett, B.; Prescott, M.F.; Solomon, S.D.; Swedberg, K.; Packer, M.; McMurray, J.J.; Shi, V.; Lefkowitz, M.; et al. Effects of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients with HFrEF. J. Am. Coll. Cardiol. 2019, 73, 795–806.
- Martens, P.; Nuyens, D.; Rivero-Ayerza, M.; Van Herendael, H.; Vercammen, J.; Ceyssens, W.; Luwel, E.; Dupont, M.; Mullens, W. Sacubitril/valsartan reduces ventricular arrhythmias in parallel with left ventricular reverse remodeling in heart failure with reduced ejection fraction. Clin. Res. Cardiol. 2019, 108, 1074–1082.
- de Diego, C.; González-Torres, L.; Núñez, J.M.; Inda, R.C.; Martin-Langerwerf, D.A.; Sangio, A.D.; Chochowski, P.; Casasnovas, P.; Blazquéz, J.C.; Almendral, J. Effects of angiotensin-neprilysin inhibition compared to angiotensin inhibition on ventricular arrhythmias in reduced ejection fraction patients under continuous remote monitoring of implantable defibrillator devices. Heart Rhythm. 2018, 15, 395–402.
- Rohde, L.E.; Chatterjee, N.A.; Vaduganathan, M.; Claggett, B.; Packer, M.; Desai, A.S.; Zile, M.; Rouleau, J.; Swedberg, K.; Lefkowitz, M.; et al. Sacubitril/Valsartan and Sudden Cardiac Death According to Implantable Cardioverter-Defibrillator Use and Heart Failure Cause. JACC Heart Fail. 2020, 8, 844–855.
- Curtain, J.P.; Jackson, A.M.; Shen, L.; Jhund, P.S.; Docherty, K.F.; Petrie, M.C.; Castagno, D.; Desai, A.S.; Rohde, L.E.; Lefkowitz, M.P.; et al. Effect of sacubitril/valsartan on investigator-reported ventricular arrhythmias in PARADIGM-HF. Eur. J. Heart Fail. 2022, 24, 551–561.
- Liu, X.-H.; Wang, G.-L.; Xu, Q.; Zhang, L.; Liu, H.-J. Effect of sacubitril/valsartan on the occurrence of cardiac arrhythmias and the risk of sudden cardiac death in heart failure: A meta-analysis of randomized controlled trials. Front. Cardiovasc. Med. 2022, 9, 943377.
- Pozzi, A.; Abete, R.; Tavano, E.; Kristensen, S.L.; Rea, F.; Iorio, A.; Iacovoni, A.; Corrado, G.; Wong, C. Sacubitril/valsartan and arrhythmic burden in patients with heart failure and reduced ejection fraction: A systematic review and meta-analysis. Heart Fail. Rev. 2023, 28, 1395–1403.
- Fernandes, A.D.; Fernandes, G.C.; Ternes, C.M.; Cardoso, R.; Chaparro, S.V.; Goldberger, J.J. Sacubitril/valsartan versus angiotensin inhibitors and arrhythmia endpoints in heart failure with reduced ejection fraction. Heart Rhythm. O2 2021, 2, 724–732.
- Russo, V.; Bottino, R.; Rago, A.; Papa, A.A.; Liccardo, B.; Proietti, R.; Manna, V.; Golino, P.; D’Onofrio, A.; Nigro, G. The Effect of Sacubitril/Valsartan on Device Detected Arrhythmias and Electrical Parameters among Dilated Cardiomyopathy Patients with Reduced Ejection Fraction and Implantable Cardioverter Defibrillator. J. Clin. Med. 2020, 9, 1111.
- Mujadzic, H.; Prousi, G.S.; Napier, R.; Siddique, S.; Zaman, N. The Impact of Angiotensin Receptor–Neprilysin Inhibitors on Arrhythmias in Patients with Heart Failure: A Systematic Review and Meta-analysis. J. Innov. Card. Rhythm. Manag. 2022, 13, 5164–5175.
- Weber, K.T. Aldosterone in Congestive Heart Failure. N. Engl. J. Med. 2001, 345, 1689–1697.
- Perrier, E.; Kerfant, B.-G.; Lalevee, N.; Bideaux, P.; Rossier, M.F.; Richard, S.; Gómez, A.M.; Benitah, J.-P. Mineralocorticoid Receptor Antagonism Prevents the Electrical Remodeling That Precedes Cellular Hypertrophy After Myocardial Infarction. Circ. 2004, 110, 776–783.
- Ouvrard-Pascaud, A.; Sainte-Marie, Y.; Perrier, R.; Soukaseum, C.; Cat, A.N.D.; Royer, A.; Le Quang, K.; Charpentier, F.; Demolombe, S.; Mechta-Grigoriou, F.; et al. Conditional Mineralocorticoid Receptor Expression in the Heart Leads to Life-Threatening Arrhythmias. Circulation 2005, 111, 3025–3033.
- Brilla, C.G. Aldosterone and myocardial fibrosis in heart failure. Herz 2000, 25, 299–306.
- Cittadini, A.; Monti, M.G.; Isgaard, J.; Casaburi, C.; Strömer, H.; Di Gianni, A.; Serpico, R.; Saldamarco, L.; Vanasia, M.; Saccà, L. Aldosterone receptor blockade improves left ventricular remodeling and increases ventricular fibrillation threshold in experimental heart failure. Cardiovasc. Res. 2003, 58, 555–564.
- Brown, N.J. Eplerenone: Cardiovascular protection. Circulation 2003, 107, 2512–2518.
- Bauersachs, J.; Heck, M.; Fraccarollo, D.; Hildemann, S.K.; Ertl, G.; Wehling, M.; Christ, M. Addition of spironolactone to angiotensin-converting enzyme inhibition in heart failure improves endothelial vasomotor dysfunction: Role of vascular superoxide anion formation and endothelial nitric oxide synthase expression. J. Am. Coll. Cardiol. 2002, 39, 351–358.
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717.
- Pitt, B.; Williams, G.; Remme, W.; Martinez, F.; Lopez-Sendon, J.; Zannad, F.; Neaton, J.; Roniker, B.; Hurley, S.; Burns, D.; et al. The EPHESUS Trial: Eplerenone in Patients with Heart Failure Due to Systolic Dysfunction Complicating Acute Myocardial Infarction. Eplerenone Post-AMI Heart Failure Efficacy and Survival Study. Cardiovasc. Drugs Ther. 2001, 15, 79–87.
- Bapoje, S.R.; Bahia, A.; Hokanson, J.E.; Peterson, P.N.; Heidenreich, P.A.; Lindenfeld, J.; Allen, L.A.; Masoudi, F.A. Effects of Mineralocorticoid Receptor Antagonists on the Risk of Sudden Cardiac Death in Patients with Left Ventricular Systolic Dysfunction: A meta-analysis of randomized controlled trials. Circ. Heart Fail. 2013, 6, 166–173.
- Le, H.-H.; El-Khatib, C.; Mombled, M.; Guitarian, F.; Al-Gobari, M.; Fall, M.; Janiaud, P.; Marchant, I.; Cucherat, M.; Bejan-Angoulvant, T.; et al. Impact of Aldosterone Antagonists on Sudden Cardiac Death Prevention in Heart Failure and Post-Myocardial Infarction Patients: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2016, 11, e0145958.
- Rossello, X.; Ariti, C.; Pocock, S.J.; Ferreira, J.P.; Girerd, N.; McMurray, J.J.V.; Van Veldhuisen, D.J.; Pitt, B.; Zannad, F. Impact of mineralocorticoid receptor antagonists on the risk of sudden cardiac death in patients with heart failure and left-ventricular systolic dysfunction: An individual patient-level meta-analysis of three randomized-controlled trials. Clin. Res. Cardiol. 2018, 108, 477–486.
This entry is offline, you can click here to edit this entry!