Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Fibrosis represents a process characterized by excessive deposition of extracellular matrix (ECM) proteins. It often represents the evolution of pathological conditions, causes organ failure, and can, in extreme cases, compromise the functionality of organs to the point of causing death.
- epigenetic
- inflammation
- fibrosis
- DNA methylation
1. Introduction
Fibrosis, characterized by the deposition of connective tissue in a tissue or organ, represents a reaction to an injury and has reparative or pathological significance. The fibrotic evolution of a tissue or organ can have very negative consequences, leading to the inability to perform normal physiological functions and resulting in a pathological condition with high mortality [1,2,3,4]. Fibrosis is often associated with pathologies characterized by a chronic inflammatory state, such as autoimmune diseases or tumors. In these circumstances, the prolonged release of growth factors and/or pro-inflammatory factors such as transforming growth factor-β (TGF-β) or various cytokines mediate the activation of a cellular transformation process called epithelial–mesenchymal plasticity (EMP) [5]. When EMP is activated, the epithelial cells, which have a phenotype of adherent cells closely connected to each other and are not invasive, become transformed, assuming a hybrid epithelial/mesenchymal phenotype and/or a completely mesenchymal phenotype. In this case, the process is defined as epithelial-to-mesenchymal transition (EMT) [5]. These cells acquire much higher migratory capabilities and are able to deposit extracellular matrix (ECM) proteins. The triggering of various cascades of molecular interconnected events leads to an exacerbation of the inflammatory state or to tumor proliferation and metastasis, with serious consequences [6,7,8]. Despite the fact that fibrosis appears to be a partly reversible process in various clinical studies [9], unfortunately, therapeutic options are still very limited. In recent years, very innovative studies have demonstrated how epigenetic modifications, by triggering or inhibiting gene transcription depending on the circumstances, can reprogram gene expression by adapting it to exposure to various risk factors [10]. This has been demonstrated, for example, in idiopathic pulmonary fibrosis (IPF) or in patients with non-alcoholic fatty liver disease, in which biopsy samples show higher expression of DNA methyltransferase, suggesting that DNA methylation could represent a predisposing factor for the onset of these pathologies [11]. The application of sequencing technology has demonstrated that the activation of fibroblasts, involved in collagen deposition during fibrogenesis, depends on various epigenetic modifications affecting the DNA to be transcribed [12]. Furthermore, epigenetic modifications appear to be largely involved in the modifications of epithelial cells towards the mesenchymal phenotype, a process essentially mediated by EMT [13,14].
2. The Dynamic Balance between EMT and EMP
EMT is a dynamic complex process during which epithelial cells reduce their epithelial properties, gradually dissolving cell–cell junctions and rebuilding cell–matrix connections to acquire characteristics typical of mesenchymal cells [15,16,17]. When the mechanism of EMT was identified, it was discovered that EMT was responsible for multiple processes involved in embryonic development, such as gastrulation, neural crest formation, and heart development [15,18]. But researchers soon demonstrated that the activation of EMT also affected physiological processes represented by wound healing [19], with the fibrotic evolution of diseases characterized by chronic inflammation, and with the formation of metastases from primary tumors. EMT is classified into three functional types: type I, involved in embryonic morphogenesis; type II, responsible for normal wound healing, but this type can enhance myofibroblast activation leading to the deposition of high levels of ECM proteins and fibrosis in chronic diseases; and type III, characteristic of malignant epithelial cells that acquire a migratory phenotype capable of invading and metastasizing [20,21,22]. EMT is a reversible phenomenon, and the resulting cells shift back from motile, multipolar mesenchymal types to polarized epithelial types via the mesenchymal–epithelial transition (MET) process [23]. Therefore, until now, EMT was considered as an “all or nothing” program wherein the cells can exist with an epithelial morphology or in a mesenchymal state. Interestingly, novel insights have shown that the cells that undergo to EMT present multiple intermediate phenotypes. This new concept, recently named as EMP, defines the capacity of the cells to interconvert between several states along the epithelial–mesenchymal spectrum, thereby acquiring hybrid epithelial/mesenchymal phenotypic features [24,25]. Intriguingly, this cellular plasticity is very pliable, and epithelial cells often undergo partial reorganization and combine epithelial and mesenchymal features following the EMT process [26] (Figure 1). Indeed, such cellular shifts and the resultant heterogeneity provide the cells with the flexibility to face different physiological (embryonic development, wound healing) and pathological (organ fibrosis, cancer) conditions [27]. The dynamics of EMT/EMP and MET are controlled by a complex network of transcription factors (TFs) [28]. TFs, in epithelial cells, determine the transcription of a variety of genes involved in the activation of EMT programs [29]. These changes in transcription, sometimes seen as gene reprogramming, involve three TFs families: Snail (Snail1) and Slug/Snail2, ZEB1 and ZEB2, and Twist [28,30]. All of these TFs share the ability to repress epithelial genes like the E-cadherin encoding gene CDH1 via binding to E-Box motifs in their cognate promoter regions [15]. In parallel, the EMT-TFs, directly or indirectly, activate genes associated with a mesenchymal phenotype, including VIM (Vimentin), FN1 (Fibronectin), and CDH2 (N-cadherin) [15,28]. Upon induction of an epithelial plasticity response, they are considered as “master” drivers of the EMT program, conferring cellular shift among the epithelial–mesenchymal spectrum [26,29]. Interestingly, with increasing data relating to the mechanisms of activation of EMT pathways, other than the signaling molecules regulating EMT, it becomes clear that activation and execution of EMT occur as a result of genetic and epigenetic processes. The study of epigenetic regulation is an important aspect of modulation of EMT [25,31], and various epigenetic mechanisms appear to be involved in the modulation of EMT, although it is still difficult to correlate all of the scientific data collected together [25]. Currently, most of the studies carried out concern the epigenetic control of EMT during cancer progression and metastases formation [16,17,18]. Similarly, recent discoveries have also attributed a key role to epigenetic modifications in the activation of the EMP process. Numerous pieces of evidence have demonstrated an altered expression of the main epigenetic modifications underlying the delicate balance between EMP and EMT, including histone modification, DNA methylation, and non-coding RNA, which could facilitate cancer metastasis [31].
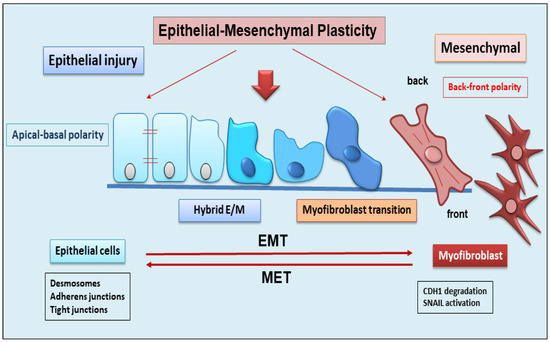
Figure 1. Schematic representation of epithelial to mesenchymal plasticity (EMP). EMP, in response to epithelial injury, allows cells to convert between multiple states across the epithelial to mesenchymal transformation, acquiring hybrid epithelial/mesenchymal phenotypic features.
3. Role of EMP/EMT in Organ Fibrosis
In recent years, our knowledge of the fibrotic process has been remarkably increased by the characterization of cellular mediators, key inflammatory and profibrogenic cytokines, molecular factors, and the evolution of new pathogenetic scenarios. A major determinant of fibrosis is the continuous spread of fibroblasts and myofibroblasts, which suggests the question of how this cellular system can be fed [32]. Experiments conducted in the last 2 years have shown that cellular plasticity, which also includes the phenomenon of EMP, is not limited exclusively to development; it also characterizes cells that undergo reprogramming that occurs during the repair of tissue damage, during fibrotic processes, and during carcinogenesis [26]. However, knowledge of the molecular mechanisms involved in the cell’s ability to modify its phenotype by evolving into another cell type is still at the beginning. The mechanisms underlying EMT are much more explored and known, and numerous studies have been conducted to evaluate the key role of EMT in fibrosis. In the context of identifying cellular drivers of fibrosis, various in vitro and in vivo studies have reported that EMT is a key mechanism during fibrogenesis, substantially contributing to the increase in interstitial fibroblasts and myofibroblasts, and interrupting its progression can have a profound impact on the onset and progression of related diseases, particularly fibrosis [21,33,34]. In fact, a fibrotic evolution of chronic diseases can lead to pathological states affecting various organs, including the lungs, liver, kidneys, heart, and salivary glands [35].The following paragraph summarizes the most recent discoveries derived from evaluating the molecular mechanisms underlying the process of EMT-dependent fibrosis in various organs in pathological conditions in order to identify potential therapeutic targets.
Contribution of Epithelium to the Fibrotic Organ Process via EMT Activation
In the last few years, important findings have demonstrated that liver epithelial cells undergo the EMT process, contributing to their transformation into myofibroblasts. Indeed, hepatocytes in which the Snail1 gene was deleted by using Cre-loxP technology showed a reduction in EMT factors and a decrease in the severity of the inflammatory response compared to controls [36]. Moreover, Rowe et al. have examined a panel of genes known to contribute to the progression of liver fibrosis, including interstitial collagen types I and III and fibroblast markers, demonstrating that Snail1 determines an increased expression of profibrotic genes such as those encoding for type I or type II collagen, vimentin, and FSP1 in the liver [36]. These data demonstrate that the hepatocyte Snail1 gene is a potent inducer in the progression of hepatic fibrosis. Indeed, explorations have been performed to determine the origin of hepatic myofibroblasts activated in response to the type of liver injury. In particular, hepatic stellate cells (HSCs) are capable of transforming into contractile myofibroblasts after liver injury. In a mouse model subjected to hepatotoxic CCL4 liver injury, activated HSCs transformed almost totally into myofibroblasts, whereas cholestatic bile duct ligation treatment preferentially stimulated portal fibroblasts [37,38]. It is interesting to underline that HSCs, when not activated, predominantly express epithelial markers compared to mesenchymal ones, and, following damage, can become activated and undergo a change in phenotype driven by EMT [39].
Nowadays, it has been experimentally proven that the activation of an EMT program occurs in a variety of pulmonary fibrosis diseases [40]. A study highlighted the contribution of the bronchial epithelial cells that, when treated with TGF-β1, are able to acquire myofibroblast phenotypes, thereby leading to peribronchial fibrosis. This process drives airway epithelium remodeling, which is a feature of asthma [41]. In IPF, alveolar epithelial cells undergo EMT, inducing the formation of fibroblastic foci and thus triggering the fibrotic destruction of the lung architecture [42,43]. Interestingly, pleural mesothelial cells also undergo a special type of EMT, mesothelial-to-mesenchymal transition (MMT), during IPF pathogenesis. In the MMT process, the mesothelial cells, during serosal inflammation, acquire the mesenchymal phenotype and complete their transformation into myofibroblasts, thus contributing to the progression of parenchymal fibrosis that results in a progressive decline in lung function [44]. Indeed, an important report demonstrated the presence of pleural mesothelial cells exhibiting mesenchymal markers in the lung parenchyma of patients with IPF after fibrogenic stimulation in vivo and a correlation between disease severity and the degree of fibrosis [44].
It has now been widely demonstrated that tubular epithelial cells (TECs) are involved in a process of EMT-dependent fibrosis in chronic renal failure [45]. However, the percentage of TECs that transform into myofibroblasts during this process is not yet known. This has led to the proposal that renal epithelial cells would undergo a partial EMT (pEMT), resulting in renal fibrotic evolution [46,47]. During pEMT, TECs maintain some characteristics of epithelial cells and show, at the same time, typical markers of fibroblasts, acquiring an intermediate phenotype between the two cell types [46,47]. Therefore, TECs in this partial mesenchymal and epithelial phenotype remain attached to the basement membrane during the fibrotic process. Recently, it was demonstrated that Snail1 is able to trigger the pEMT process in TECs, relaying crucial signals for fibrogenic cytokine release and promoting differentiation into myofibroblasts, thus contributing to the exacerbation of the inflammatory response [46].
EMT is also activated in inflammatory bowel diseases (IBDs) such as ulcerative colitis (UC) and Crohn’s disease (CD) [48]. In IBD patients, persistent intestinal inflammatory factors injure intestinal epithelial cells, determining reparative reactions that lead to the triggering of the EMT process and perpetuating a severe fibrotic condition [48,49]. Confirming this, the presence of high levels of tumor necrosis factor-like ligand 1A (TL1A) in the intestinal specimens of patients with UC and CD has been detected, which represents a potent inducer of EMT in intestinal fibrosis. As expected, the TGF-β1/Smad3 pathway may be involved in TL1A-induced EMT [49,50,51].
Recent discoveries have also shown that uncontrolled fibrosis is present in the heart and is triggered by EMT and its special type, endothelial to mesenchymal transition (EndMT) [52,53]. Epicardial EMT is activated after myocardial infarction, atherosclerosis, and valve dysfunction, and it determines angiogenesis and healing [54]. Under TGF-β stimuli, cardiac fibroblasts transdifferentiate into myofibroblasts, acquiring a phenotype similar to that of smooth muscle cells. Furthermore, fibroblasts can also originate from endothelial cells through EndMT, giving rise to the progression of cardiac fibrosis [54]. In addition, TGF-β-driven EMT responsible for cardiac fibroblast formation appears to be triggered by the Hippo pathway, an evolutionarily conserved kinase cascade [55], as seen in recent in vitro findings [56]. In addition, the process of cardiac fibrosis seems to be regulated by C-Ski protein, identified as an inhibitory regulator of TGF-β signaling [57].
A flourishing research field is focused on the evaluation of the pathways involved in the fibrotic process observed in the salivary glands (SGs) derived from Sjögren’s Syndrome (SS) patients. Fibrogenesis observed in SGs can be considered the end result of chronic, intense inflammatory reactions induced by a variety of stimuli in this autoimmune disease [7,8,58,59]. Pioneering studies aimed at correlating SS with a fibrotic evolution of the salivary glands were conducted over 10 years ago, demonstrating a significant association between stimulated salivary flow, the focus score, and fibrosis in a high number of SS biopsy specimens. In comparison, unstimulated salivary flow appears to be weakly associated with the focus score and is not always correlated with fibrosis, which was considered an excellent measure of irreversible damage [58]. In all cases, SG fibrosis is linked with an evident impairment of organ function that leads to progressive atrophy and a decrease in quality of life for patients [60].
In this context, using technology to create transgenic mice that conditionally overexpress active TGF-β1, experimental data have confirmed that the overexpression of active TGF-β1 leads to an abnormal accumulation of ECM proteins and severe hyposalivation and acinar atrophy in the mutated mice [61]. More recently, studies have demonstrated an exuberant upregulation of TGF-β1 in SS SGs, which induces the loss of epithelial features and the acquisition of mesenchymal features in SG epithelial cells via triggering of the EMT program through the TGF-β1/Smad/Snail signaling pathway [62]. Indeed, TGF-β1 seems to be able to regulate EMT through both main pathways: the canonical Smad-dependent and non-canonical Smad-independent signaling pathways [7,8,62].
4. Main Epigenetic Mechanisms
Each phase of gene expression can undergo epigenetic modifications, thus leading to the synthesis or inhibition of certain downstream proteins. The epigenetic processes involve DNA methylation, histone modification, chromatin remodeling, and the effects of noncoding RNA.
The phenomenon of DNA methylation is an essential process for the physiological development of the individual and plays a key role in processes widely studied in recent years, such as genomic imprinting. DNA methylation and demethylation represent heritable epigenetic signatures that are evolutionarily conserved and do not involve an alteration of the DNA sequence but can, however, lead to widely modified gene expression [63,64].
Methylation of DNA is an epigenetic mechanism that consists of the transfer of a methyl group from S-adenyl methionine (SAM) to the C-5 position of a cytosine residue in a dinucleotide CG or polynucleotide CGGCGG context, also termed CpG islands, to form 5-methylcytosine catalyzed by DNA methyltransferases (DNMTs) [65]. The DNMT family comprises various elements: DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L [66]. Notably, the methylation of the promoter region or gene has different effects; excess promoter methylation silences the gene, while a reduction in promoter methylation causes increased gene expression [67]. On the contrary, at the gene level, an excess of methylation determines active transcription of the gene itself, while the methylation of the gene has a meaning that is not yet well known [68].
Similar to DNA methylation, post-translational histone modifications do not affect the DNA nucleotide sequence but alter its accessibility to the transcriptional machinery. Histones are small basic proteins assembled into nucleosomes and are essential to compact and stabilize DNA by making the DNA sites implicated in gene transcription accessible [69]. Each nucleosome is composed of approximately 150 base pairs of DNA and two copies of the four core histones: H2A, H2B, H3, and H4 [69]. In addition, H1 protein acts as a linker histone-compacting chromatin, and its role is to stabilize the internucleosomal DNA but does not form part of the nucleosome. Histone proteins, through post-translational modifications, control chromatin structure, triggering the transition from open chromatin, called euchromatin, which is actively transcribed, to a compacted chromatin structure called heterochromatin. In this compact form, DNA is not accessible to transcriptional machinery and thus cannot be transcribed, resulting in gene silencing [70].
Several of the best-known post-translational modifications of histones include acetylation, methylation, phosphorylation, and ubiquitylation. However, in recent years, other histone modifications have been identified, such as GlcNAcylation, citrullination, krotonylation, and isomerization, which still need to be further explored [71].
Acetylation modulates transcriptional activity through the neutralization of the positive charge present on the lysine residues of histone proteins. This action has the potential to weaken the interactions between histones and DNA, making them less stable, thus allowing gene transcription [72,73]. Acetylation consists of the addition of acetyl groups to lysine residues, neutralizing their positive charge. Thus, acetylation induces and enhances gene expression. Histone acetylation and deacetylation are catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively.
HDACs remove acetyl groups from acetylated proteins, consequently repressing gene expression. They are classified into four classes: class 1 (HDAC1,2,3,8), class 2 (2a: HDAC4,5,7,9; 2b: HDAC6,10), class 3 (SIRT), and class 4 (HDAC11). Therefore, sirtuin proteins, classified within class III HDACs, require nicotinamide adenine dinucleotide (NAD) as a cofactor for their catalytic activity. To date, 18 mammalian HDACs have been identified and classified into the above different classes [74].
Methylation occurs in both the lysine and arginine residues of histones H3 and H4 and, in particular, does not alter the charge of the histone protein. Arginine methylation, which requires arginine methyltransferase activity, induces gene transcription, while lysine methylation, which requires histone methyltransferase, can have either a positive or negative effect on transcription due to the site involved in the methylation [75,76,77,78]. Recently, it has been demonstrated that histone methylation is also a reversible event through the mechanism of histone demethylases [79].
Phosphorylation influences all core histones, with several effects on each. Phosphorylation of serine residues 10 and 28 of histone H3 and serine residue T120 of histone H2A is involved in chromatin condensation through the phases of cell replication during mitosis. Phosphorylation of the S139 residue in histone H2A evidences a landing point for the interaction with factors involved in the repair of DNA damage [80]. However, phosphorylation of histone H2B is not as well known but appears to induce chromatin compaction through several mechanisms such as apoptosis, DNA fragmentation, and cell necrosis [81].
All histone proteins can undergo a mechanism of ubiquitylation; however, in the last few years, studies have highlighted two well-characterized proteins, H2A and H2B, which are most frequently ubiquitinated in the nucleus [82]. Histone ubiquitination is linked with the activation of gene expression, but many studies have demonstrated that the presence of a single ubiquitin has different effects on H2A and H2B. Indeed, mono-ubiquitylated H2A is linked with gene silencing, while if the interaction concerns H2B, transcription activation is induced [82].
Epigenetic regulation also involves actively non-coding RNA (ncRNAs) and it has been widely discovered that ncRNAs are able to modulate gene expression at both transcriptional and post-transcriptional levels. ncRNA refers to a functional RNA molecule that is transcribed from DNA but is not translated into a protein [83]. NcRNA are divided into two broad categories based on their length: short ncRNAs, with a number of nucleotides less than 30, and long ncRNAs (lncRNAs), which include those RNAs with a number of nucleotides greater than 200 [83]. The three main classes of short noncoding RNAs include microRNAs (miRNAs), short interfering RNAs (siRNAs), and piwi-interacting RNAs (piRNAs).
MiRNAs and DNA methylation are the two epigenetic events that have emerged in recent years and correlate to the modulation of gene expression [84]. Notably, miRNAs act by linking to a specific target messenger RNA through a complementary sequence; this binding determines the fragmentation and degradation of the mRNA, consequently blocking the translation event. Interestingly, the presence of a mutual regulation between miRNAs and DNA methylation has been shown in human tumors. Indeed, miRNAs modulate DNA methylation by acting on the transcription of genes implicated in the synthesis of DNA methyltransferases [85].
SiRNAs represent small RNA molecules whose function is to repress the expression of a gene by binding to the mRNA, inducing its degradation, and thus preventing post-transcriptional gene and subsequent protein synthesis [85]. SiRNAs are gained from a long double-stranded RNA molecule that is cut into many small fragments by Dicer endoribonuclease. The siRNAs obtained are added to the so-called RISC complex (RNA-induced silencing complex) to form the inactive RISC-siRNA complex. Once activated, the siRNA loses one of the two strands and binds to the mRNA target messenger, i.e., the mRNA whose translation into protein is to be prevented [86,87]. The piRNAs are a complex class of sncRNAs that specifically interact with the PIWI protein subfamily of the Argonaute family [88]. Current research evidences that this interaction between PIWI proteins and piRNAs regulates novel epigenetic mechanisms such as DNA rearrangements; however, this has yet to be clarified. Recently, lncRNAs were discovered as important regulators of the epigenetic status of the human genome. LncRNAs are RNA fragments longer than 200 nucleotides that have various activities, such as chromatin remodeling and transcriptional and post-transcriptional regulation, and act as precursors of siRNAs with the function of gene silencing [89,90,91]. Many lncRNAs form complexes with proteins, leading to modifications in the conformation of chromatin [92,93]. In addition, a novel member of the lncRNA class, circular RNAs (circRNAs), are characterized by a covalently closed loop. They are recognized to have distinct biogenesis and to regulate gene expression and biological processes through different mechanisms, with some miRNA-sponging circRNAs identified [94]. A schematic overview of epigenetic modifications is reported in Figure 2.
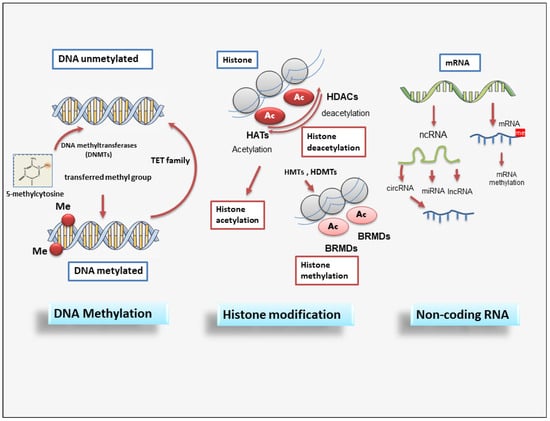
Figure 2. A schematic overview of epigenetic modifications. Epigenetic regulation involves DNA methylation, histone modification, and non-coding RNAs. DNMT family members mediate DNA methylation, which suppresses gene transcription by adding a methyl group to the cytosine position. Histone methylation is catalyzed by HMTs and HDMTs, and histone acetylation is regulated by HATs and HDACs. Non-coding RNAs include miRNAs, circRNAs, and lncRNAs. BET (Bromodomain and extraterminal); BRMDs (bromodomains); DNMT (DNA methyltransferase); HATs (Histone acetylases); HDACs (Histone deacetylases); HDMTs (Histone demethylases); HMTs (Histone methyltransferases); lncRNA (long non-coding RNA); TET (Ten-eleven translocation).
This entry is adapted from the peer-reviewed paper 10.3390/ijms25052775
This entry is offline, you can click here to edit this entry!