Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Coccidioidomycosis is an important fungal disease that is found in many desert regions of the western hemisphere.
- coccidioidomycosis
- immunity
- innate immunity
- macrophages
1. Introduction
Coccidioides spp. are highly pathogenic organisms but only 50% of immunocompetent hosts develop symptomatic disease. A large majority of pulmonary infections are self-limited and often go undiagnosed. Thus, the human immune system is usually able to control the infection with either no residual evidence of prior infection or a solitary pulmonary nodule [1,2,3]. For this reason, evaluating mammalian immunity is critical for an understanding of the host–pathogen relationship. Coccidioides species are dimorphic fungi. Their life cycle consists of a saprobic phase characterized by mycelia that produce enterothallic arthroconidia and a parasitic phase characterized by endosporulating spherules. The parasitic cycle of Coccidioides is unique amongst the medically important fungi and the ability to form spherules from arthroconidia is required for pathogenicity. Fundamental research on their genetic, morphology, ecology, virulence factors, and pathogenic mechanisms has recently been reviewed [4,5].
2. Innate Immunity
There are many reviews of innate immunity in coccidioidomycosis [2,6,7,8,9]. An especially informative and complete review of this topic has recently been published [10]. The innate immune response to infectious organisms plays a key role in slowing the course of the infection while the host is generating an antigen-specific immune response, and the innate response influences the type of adaptive immune response that is made by virtue of the cytokines and chemokines produced by innate immune cells [11].
2.1. Local Factors in the Lung
Pulmonary epithelial cells interact with the arthroconidia very early in the course of infection. There have been some fascinating hypotheses about the importance of this interaction, but no data have been published [12]. Recently, Hsu reported that mutations in Duox1, a lung epithelial cell oxidase that generates H2O2, were more common in patients with disseminated coccidioidomycosis than in the general population, suggesting that this enzyme plays a role in innate resistance to coccidioidomycosis [13]. Whether arthroconidia or round cells are killed by physiological concentrations of hydrogen peroxide is not known, but there is some evidence that they are relatively resistant to oxidative stress [14]. Pulmonary surfactant is also an early host fluid to encounter arthroconidia in the lung. It has been reported that mice challenged with C. posadasii arthroconidia had dramatically lower concentrations of SP-A and SP-D in their bronchoalveolar fluid (BALF) compared to controls 10 days after challenge [15]; immunization of mice before the challenge prevented this effect. It is not clear whether the alterations in surfactants occur early in infection or are a consequence of the development of pneumonia. The roles of epithelial-associated myeloid cells, such as NK and iNKT cells, and epithelial-associated dendritic cells in innate resistance to pulmonary coccidioidomycosis, are unknown.
2.2. Polymorphonuclear Leukocytes
Polymorphonuclear leukocytes (PMN) are cells that act early in the course of infection, phagocytosing and inhibiting the growth of arthroconidia to some degree [16]. There is ample histologic evidence that collections of PMNs occur in pathologic lesions, although the original arthroconidia at this point in infection have either died or converted to spherules (Figure 1).
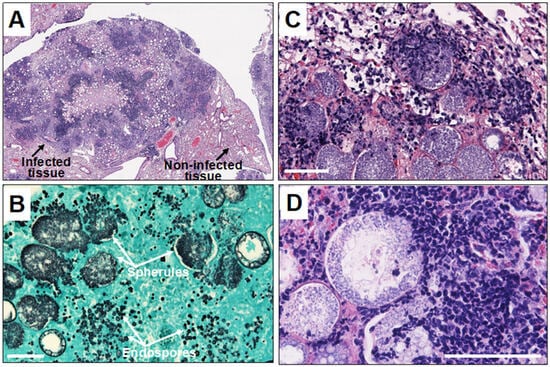
Figure 1. Histopathology of Coccidioides infection in mice. Legend: (A) microscopic image of Coccidioides-infected mouse lung tissue showing disorganized pyogranuloma. Empty circles are large spherules with central vacuoles in the infected tissue; (B) large spherules and tiny endospores in the infected tissue stained with Gomori-methenamine silver; and (C,D) microscopic images showing infiltration of neutrophils and other granulocytes toward endosporulating spherules. The white scale bar represents 100 µm.
Chemotaxis is required for PMN to move from the circulation to the organism in tissues. In chemotactic assays using mycelial or spherule culture filtrate, complement was found to be required for the chemotactic response [16]. A recent study using very sophisticated imaging techniques found that human PMN recognized spherules and endospores over a distance of several micrometers in the presence of serum [17]. The PMN then phagocytosed both spherules and endospores in the presence of human serum containing active complement.
Human PMNs inhibit chitin synthesis by arthroconidia in vitro, especially in the presence of complement [18]. This inhibition lasted more than six, but less than 24 hours. PMNs from a patient with chronic granulomatous disease did not inhibit chitin synthesis. However, PMN from normal donors did not kill Coccidioides spp. over the first 18 h of co-incubation, despite phagocytosis of the arthroconidia and degranulation of the PMNs. Inhibition of chitin synthesis by PMNs or H2O2 was also observed in small (young) spherules but not in large (mature) spherules [19]. Another study found that both young and mature spherules were phagocytosed very poorly by PMN’s compared to arthroconidia, spherule initials, or endospores [20]. Very few organisms were killed, although there were some differences between the four clinical isolates that were studied. In most cases, phagocytosed endospores did not elicit degranulation of the PMNs, which is consistent with the minimal killing observed. Segal reported that a variety of cationic peptides were able to inhibit the growth of arthroconidia [21]. Studies of cationic peptides with spherules and endospores have not been reported.
Studies in IL-8 Receptor 2 knockout (IL-8R2−/−, also known as CXCR2−/−) mice on a BALB/c background suggest that PMN’s may not play a protective role in experimental mouse infection [22]. Although mice do not produce IL-8, they do make the chemotactic factors MIP-2 and KC, which are ligands for this receptor in mice. Normal mouse macrophages stimulated with spherules make MIP-2. As expected, fewer PMN’s were found in the lungs of C. immitis-infected IL-8R2−/− mice compared to BALB/c controls. Unexpectedly, the IL-8R2−/− mice had 10-fold lower fungal quantitative culture values (CFU) in their lungs and spleens 14 days after infection compared to the controls. Survival studies were not conducted.
To evaluate the molecular mechanisms behind increased resistance to this infection, RNA-seq studies of mouse genes in the infected lungs were conducted. Upregulated genes in control animals were enriched for gene ontology categories related to leukocyte activation, such as inflammatory response, leukocyte migration, and neutrophil degranulation, consistent with the higher numbers of neutrophils in their lungs. Expression of IL-17, 1L-12, and interferon-γ (IFN-γ) genes, which are protective cytokines, were upregulated in the IL-8R2−/− mice. In contrast, IL-1β, TNF-α, IL-6, and the inhibitory cytokine IL-10 were more upregulated in control lungs [22]. The difference in cytokine profiles provide a rationale for the decreased fungal burden in IL-8R2 KO mice.
In contrast to those results, depletion of PMN by treatment with a monoclonal antibody did not change the severity of infection (as measured by CFU in lungs and spleens) in C57BL/6 mice [23]. The discrepancy between that result and Carlin’s results could be due to the difference in methods for eliminating the PMN response, the mouse strains used, and/or the more severe disease caused by the C. posadasii C735 infection compared to the less pathogenic C. immitis RS organism.
2.3. Monocytes/Macrophages and Dendritic Cells
The biology of monocytes, macrophages, and dendritic cells is very complex and play variety of roles in innate immunity with overlapping functions. It is important to remember that, while macrophages play an important role in the control of all types of infections, monocytes mature into different types that differ in function [24,25]. Dendritic cells are also a complex group of cells involved in sensing infection and, producing cytokines and chemokines that shape the acquired immune response, and they are the most efficient antigen presenting cells to T-lymphocytes [26].
A few older studies have reported that activation of mouse peritoneal macrophages by immune spleen cells resulted in increased phagolysosome fusion and the killing of arthroconidia and endospores [27]. A subsequent study found that peritoneal macrophages incubated with supernatants of antigen-stimulated Coccidioides-immune spleen cells also fused the phagolysosome after phagocytosis of arthroconidia. The addition of these supernatants to macrophages increased the killing of arthroconidia [28]. To our knowledge, these observations have not been reproduced by others.
A study about the interaction of arthroconidia with C57Bl/6 mouse macrophages and dendritic cells was recently published [29]. The avirulent C. posadasii cts2/cts3 deletion mutant was used to evaluate phagocytosis of arthroconidia by mouse macrophages and dendritic cells, a mouse macrophage cell line, and a rat pulmonary macrophage cell line. None of these cell types phagocytosed arthroconidia efficiently; only about 10% of the cells phagocytosed arthroconidia. Incubation of mouse bone-marrow-derived monocytes with arthroconidia resulted in differentiation into M0 macrophages (potentially immunosuppressive), but not into M1 (inflammatory) or M2 (anti-inflammatory) macrophage phenotypes, as characterized by cell surface markers. Incubation of dendritic cells with arthroconidia caused little cellular activation and was biased to the DC1 phenotype (which activates CD4 T-cells poorly) [26]. It would be informative to conduct these experiments with spherules as well as arthroconidia, and with other Coccidioides spp. and inbred mouse strains.
2.4. Cellular Receptors and Pathways for the Innate Immune Response
2.4.1. C-Type Lectin Receptors
Dectin-1, (the β-glucan receptor that is expressed on myeloid cells and some lymphocytes) [30] was first shown to be required for a mouse macrophage cell line to produce pro-inflammatory cytokines when stimulated with inactivated spherules or β-glucan isolated from spherules. Cells that did not express Dectin-1 were unresponsive to those stimuli and an anti-Dectin-1 monoclonal antibody substantially reduced the pro-inflammatory cytokine response of mouse peritoneal elicited macrophages to spherules. Therefore, Dectin-1 appears to be an important receptor for the pro-inflammatory cytokine response of murine peritoneal macrophages to spherules. Furthermore, elicited peritoneal macrophages from C57BL/6 Dectin-1−/− mice made much less TNF-α, IL-6, and MIP-2 in response to spherules than the peritoneal macrophages from control C57BL/6 mice [31]. However, the response of bone-marrow-derived dendritic cells (BMDC) to spherules was somewhat different. The IL-6, IL-10, and granulocyte-macrophage colony-stimulating factor (GM-CSF) responses to spherules were mostly dependent on Dectin-1, but IL-1 and TNF-α production was independent of this receptor. The course of infection was also influenced by Dectin-1. C57BL/6 Clec7a−/− mice (lacking Dectin-1) had significantly more organisms in their lungs and substantially more organisms in their spleens compared to the controls, suggesting that the activation of Dectin-1 reduces dissemination from the lungs [32]. In addition, bronchial alveolar lung fluid (BALF) from Dectin-1−/− mice obtained after intra-nasal infection contained lower concentrations of protective TH17 and TH1 cytokines than BALF from infected control mice. TH17 cytokines are required for the successful immunization of mice to Coccidioides spp. with an attenuated live vaccine [33], although the role of TH17 cytokines in human coccidioidomycosis is not yet established.
Murine susceptibility to experimental coccidioidomycosis is greatly influenced by the genetic background of the animal: C57BL/6 mice are relatively susceptible to infection and DBA/2 are relatively resistant [34]. The two strains of mice had the same amount of Dectin-1 in macrophages, but the Dectin-1 transcript was about 100 bp shorter in the C57BL/6 mice than in the DBA/2 mice. The truncated Dectin-1 lacked the extracellular stalk, which was the result of alternative splicing of clec7a by C57BL/6 mice that eliminated the expression of exon 3 [35]. C57BL/6 x DBA/2 recombinant inbred (BXD RI) mice with the truncated form of Dectin-1 were more susceptible to C. immitis infection than the RI mice that expressed the full-length protein [31]. A cell line transfected with the truncated C57BL/6 isoform of Dectin-1 made half as much TNF-α as those transfected with full-length Dectin-1 when stimulated with killed spherules. This is the opposite of how the two splice variants affect macrophage responses to C. albicans [35]; therefore, these two organisms elicit different responses to the truncated protein. In contrast to these studies, Campuzano et al. did not find that Dectin-1−/− C57BL/6 mice were more susceptible to a primary infection with C. posadasii C735 than C57BL/6 controls, although vaccination of the mutant mice with a live-attenuated vaccine (ΔT strain) did not protect Dectin-1−/− mice as well as the controls [33]. This may be because the infection in unvaccinated C57BL/6 mice was so severe that it was not possible to detect the immune defect caused by the absence of Dectin-1 [33]. However, vaccination of Dectin-1−/− mice with a different live-attenuated vaccine (Δcps1) was relatively protective against infection with C. posadasii Silveira [36]; so the role of Dectin-1 in vaccine-induced immunity is unclear.
The roles of murine Dectin-2 and the mannose receptor (MR) on cytokine secretion in vitro and resistance to infection in vivo have also been studied. BMDC from C57Bl/6 MR−/− mice make less pro-inflammatory cytokines in vitro in response to spherules compared to controls, even though the MR receptor is not a signaling receptor [37]. However, MR−/− mice are not more susceptible to C. immitis infection [37]. Furthermore, the mannose receptor plays a role in the recognition of C. posadasii spherules by human DCs, since spherule binding and cytokine production can be inhibited by mannan [38,39]. The discrepancy between the effect of the MR on vitro response and the course of infection is unexplained.
Peritoneal macrophages from Dectin-2−/− mice made poor pro-inflammatory cytokine responses to spherules in vitro. Dendritic cells (BMDC) from Dectin-2−/− mice made identical protective cytokine responses in vitro to spherules, but lower amounts of IL-10. The Dectin-2−/− deletion had no effect on susceptibility to C. immitis infection [37]. Mice lacking both MR and Dectin-2 were not more susceptible to infection, indicating that these receptors did not play a critical role in host defense to coccidioidomycosis in mice, despite the role they play in vitro.
Dectin-2 does play a role in the acquired immune response to Coccidioides spp. [33,40]. Immunization of Dectin-2−/− mice with a live, attenuated mutant led to lower levels of IL-17 as well as IFN-γ. Immunized Dectin-2−/− mice were more susceptible to infection than immunized controls. Vaccine protection was dependent on the production of TH17 cells, and very few TH17 cells were present in the lungs of vaccinated Dectin-2 KO mice after infection. CARD-9 is an intracellular signal transduction adaptor molecule that is downstream of Dectin-1 and Dectin-2. CARD-9−/− mice were not protected by vaccination.
2.4.2. Toll-Like Receptors (TLR)
Elicited peritoneal macrophages from C57Bl/6 TLR-2−/− mice made a very poor pro-inflammatory cytokine response to spherules in vitro compared to macrophages from control mice [41]. However, BMDC from both MyD88−/− and MyD88/TRIF−/− mice made as much TNF-α, IL-6 and IL-10 in response to spherules as the control C57BL/6 mice [42]. Macrophages from TLR-4−/− mice made an identical response to spherules, compared to control mice, suggesting that TLR-4 is not an important receptor for the macrophage response to spherules. This observation is consistent with a previous study of genetically determined resistance to C. immitis in mice, in which a TLR-4 null mutation was found to have no effect on resistance to infection [34]. There are no data about the role of TLR on pulmonary macrophages in the response to spherules.
Despite the observation that MyD88 did not affect the cytokine response to spherules by BMDC in vitro, MyD88−/− and MyD88/TRIF−/− mice were more susceptible than controls to C. immitis infection. However, neither TLR-2−/− nor TLR-4−/− mice were more susceptible to infection. Since MyD88 is a signal-transducing molecule for both the IL-18 receptor and the IL-1 receptor, the role of each of these receptors on resistance to infection was studied. IL-18R−/− mice were no more susceptible to infection than control mice, but IL-1 receptor−/− mice were much more likely to develop disseminated disease. IL-1RA, part of the IL-1 family of cytokines, was present at a higher concentration than any other measured cytokine in the BALF of infected C57BL/6 mice, further evidence of the importance of the IL-1 family in pathogenesis [22]. These experiments suggest that signaling by IL-1 may play a critical role in innate immunity to coccidioidomycosis in mice. The IL-1 receptor and MyD88 have both been shown to be important for vaccine-induced immunity in mice [23]. The role of IL-1 in the human innate response to Coccidioides has not been studied.
2.4.3. In Vivo Studies of Murine Innate Immunity
Barker and colleagues have evaluated the response to pulmonary infection with 105 arthroconidia (an extremely high inoculum) using three different strains (two C. immitis and one C. posadasii) at days 1–5 after challenge [43]. The strains differed in pathogenicity: the C. posadasii strain Silveira was the most pathogenic, whereas the C. immitis strain 2006 was least pathogenic, and C. immitis R.S. was intermediate. Coccidioides spp. RNA could be detected in the lungs of mice infected with all three isolates by day 5 and the leukocyte count was also elevated by that time. The cytokine responses as measured by qRT-PCR of lung tissue showed that IL-1, IL-1R, IL-10, IL-17RA, and the IFN-γ receptor genes were all upregulated in response to all three challenges by day 5 after infection, whereas the other measured cytokines showed less consistent responses. The contents of BALFs were also analyzed by proteomic techniques: 38% (138/366) of the detected mouse proteins were found in mice challenged with all three isolates and 12–42 proteins were unique to each of the three challenges. Most of the detected proteins were not cytokines. One conclusion of this study is that there are differences in the host responses to the three challenge strains, as measured by cytokine production and the proteomics of host proteins.
Innate resistance to subcutaneous infection in mice has also been studied [44]. Subcutaneous inoculation of the organism is an unusual but well-known route of infection in human beings who are injured in desert soil. The course of this type of infection in wildtype mice was resolution of the infection over three months after inoculation. However, CARD-9−/− and MyD88−/− mutant mice in theC57BL/6 background died after the challenge, suggesting the importance of both TLR and C-type lectin signaling in this route of infection. Although the lesions contained many PMNs, PMN depletion with the antibody did not affect the fungal burden. IFN-γ, but not IL-17 was required for resistance to subcutaneous infection. In addition, the fungal burden was significantly higher in iNOS−/− mice than in the controls. Therefore, it seems that the factors determining innate resistance to subcutaneous infection are significantly different than those important for resisting pulmonary challenges.
Another paper also compared the cytokine response in BALF from mice infected with C. immitis RS or C. posadasii Silveira [45]. The two fungal strains studied elicited different cytokine profiles 10 days after infection, although there was a good deal of variation within each group. In addition, the volatile organic compound profiles in the BALF were found to correlate with cytokine profiles. Whether the mouse or the fungus produces these volatile organic compounds is unknown.
2.5. Genetically Determined Resistance to Infection and Innate Immunity in Mice
The initial studies of genetic differences in susceptibility to infection in mice were conducted using intraperitoneal (I.P.) infection with the C. immitis RS as a model [34]. In this model, mice develop intra-peritoneal granulomas and infection spread to lungs and other organs. There was a dramatic difference between susceptibility in strains; C57BL/6 and BALB/c mice were very sensitive to infection and DBA/2 mice were relatively resistant. Resistance to infection was not determined by the major histocompatibility class II molecules (H-2 locus). In F1 crosses, resistance was the dominant phenotype. All studies were conducted with female mice; male mice were more susceptible to infection. Immunization of the susceptible mice with a live, attenuated mutant protected them from infection, indicating that they could make a protective immune response to the organism. Backcross experiments suggested that a single locus might be the primary determinant of this phenotype [46]. In radiation chimera studies, resistance was conferred by the transplant of T-cells from the resistant strains. Other investigators reported that DBA/2 mice are also relatively resistant to intranasal infection while BALB/c mice are susceptible [47]. They found that the DBA/2 mice produced much more IFN-γ in their lungs, while susceptible mice produced more IL-4. Administration of IFN-γ to susceptible BALB/c mice improved their resistance to infection, whereas treatment of resistant mice with neutralizing anti-IFN-γ antibody increased their susceptibility, suggesting that these cytokines play a causative role in genetically determined resistance. There is also a study indicating that IL-12, a cytokine that stimulates a TH1 immune responses including IFN-γ, decreases the number of organisms in tissues after experimental infection [48].
Studies of cytokines in intraperitoneally infected genetically resistant (DBA/2) and susceptible (C57Bl/6) mice revealed that C57BL/6 mice made about 1000-fold more IL-10 and 10-fold more IL-4 in infected spleens and lungs than resistant DBA/2 mice after an I.P. infection [49]. In contrast, DBA/2 mice had more IL-12p40 in their lungs than did C57BL/6 mice. DBA/2 and C57Bl/6 mice made equivalent amounts of IFN-γ, IL-6, and IL-2. Most importantly, C57BL/6 mice with a deletion of the IL-10 gene were more resistant to infection than control mice and were equally resistant to infection as DBA/2 mice. C57BL/6 mice with an IL-4 deletion were somewhat more resistant to infection than control C57Bl/6 mice, but not as resistant as IL-10 deletion mice or DBA/2 mice. Therefore, IL-10 has a major influence on the course of infection in these mice.
Genetic mapping of the I.P. infection resistance phenotype was done in the BXD recombinant inbred mice available at that time [50]. Recombinant inbred mice are derived by intercrossing F1 animals (in this case C57BL/6 and DBA/2) for many generations to obtain inbred lines that are homozygous for different combinations of reassorted genes on all the non-sex chromosomes from both of the parental strains, which allows for the mapping of genes by comparison with previously mapped genes. In contrast to a previous study suggesting that a single gene determined resistance, the resistance phenotype was determined by at least two genes, one on chromosome 4 and the other on chromosome 6. The number of mapped genes was much smaller at the time than it is now, and there are many more RI BXD lines that could be tested, which could increase the likelihood of identifying more resistance loci/genes.
The expression of mouse genes in the lungs of resistant and susceptible infected mice was compared using microarray technology [52]. Figure 2 shows the gene expression network resistance of DBA/2 mice versus susceptible (C57BL/6) mice on day 14 after intranasal infection. Many interferon-stimulated genes, cytokines, as well as histocompatibility genes were preferentially expressed in the genetically resistant mouse strain. Upregulated transcription factors in the resistant mouse strain included hypoxia inducible factor 1, interferon regulatory factor 1, and STAT1. These results indicate that a wide variety of genes are differentially expressed in the lungs of susceptible and resistant mice after infection, but they do not indicate which, if any, of these differences are most important for resistance, nor what cells are using those transcription factors to produce the cytokines [52].
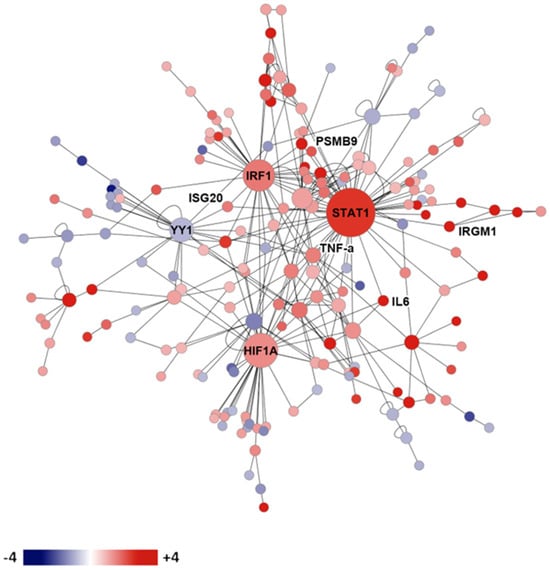
Figure 2. Gene expression network in infected genetically susceptible and resistant mice. Legend: protein expression network of gene transcription in resistant DBA/2 versus susceptible C57BL/6 mice on day 14 after infection. Red shading indicates more expression in DBA/2 than C57BL/6, while blue shading indicates the reverse.
2.6. Genetic Predisposition to Severe Disease in Human Beings
A small number of people with disseminated coccidioidomycosis have been found to have Mendelian mutations in the IL-12/IFN-γ pathway [53,54], evidence for the importance of IFN-γ and CD4 TH1 cells in immunity to coccidioidomycosis. A gain-of-function mutation in the STAT1 IFN-γ/IL-12 signaling protein that dysregulates signaling has also been observed in two patients with disseminated disease [55]. Recently, a genomic study of 58 patients with disseminated coccidioidomycosis found variants in genes coding for the IL-12/IFN-γ-signaling pathway, the innate immune-signaling pathway and the NF-κB- and IL-17-signaling pathways [6]. One variant found in a family with three generations of disseminated coccidioidomycosis was a missense mutation in STAT4 [36]. To further investigate this variant, this mutation was introduced into the mouse genome and resistance to infection was evaluated in mice infected with C. posadasii strain 1038 (a lower virulence clinical isolate that causes indolent infection in mice) [56]. The mutant mice became moribund much more quickly than the controls, indicating that this missense mutation has a dominant negative effect on the course of the disease. By contrast, C57BL/6 mice with only one1 STAT4 gene completely knocked out survived as long as the controls, which indicates that the E626G point mutation in STAT4 is dominant negative and suggests that only a single copy of the gene is required to have an observed effect on susceptibility to severe coccidioidomycosis in humans. [56] As expected, T-lymphocytes from C57BL/6 Stat4E626G/+ mice made less IFN-γ in response to IL-12 and IL-18. The number of T-lymphocytes in the mediastinal lymph nodes was also lower in the infected mutant mice than in the controls.
All these mutations have a profound effect on TH1 or TH17 immunity, which are critical host responses. People who have low numbers of T-cells or poor T-cell function because of infections, such as HIV/AIDS, immunosuppressive malignancies, or immunosuppressive medical therapies, are also at a high risk for severe infection and/or disseminated infection [2,57,58].
There is also a marked association with ethnicity with the course of human infection. African Americans are significantly more likely to develop disseminated and/or lethal disease than are those of European descent [59,60,61,62,63]. Filipinos and Filipino Americans are also much more likely to develop disseminated disease than people of European descent [61]. There are some suggestions in the literature that Hispanics are more likely to develop severe or disseminated disease, but most experts think this is unproven. The risk of infection seems to be the same in African Americans and people of European descent, and although it is difficult to exclude sociological and environmental factors, the difference in susceptibility to dissemination rather than infection has been seen in many studies in the military and in prisons, where one would expect nutrition and housing to be similar for all those exposed.
One study has correlated the ABO blood groups and some HLA alleles with the severity of infection in three broad ethic groups: Caucasians, African Americans, and Hispanics [64]. There was evidence of a linkage of severity of disease to blood group antigens only in the Hispanic patients. However, HLA DQB1 alleles were associated with decreased or increased risk in all three populations.
A recent study has conducted extensive genetic analysis of people with disseminated coccidioidomycosis using whole-exome sequencing [13]. This approach makes no assumptions about the polymorphisms that might be associated with disseminated disease. They studied 67 patients in an exploratory set and 111 patients in a validation set. In the exploratory set, 2 patients had STAT3 mutations and 34 had defects in the β-glucan sensing pathway. These mutations included point mutations in the clec7a gene coding for Dectin-1 and a PLCG2 variant coding for phospholipase C γ-2, which is activated via Dectin-1 (Figure 3).
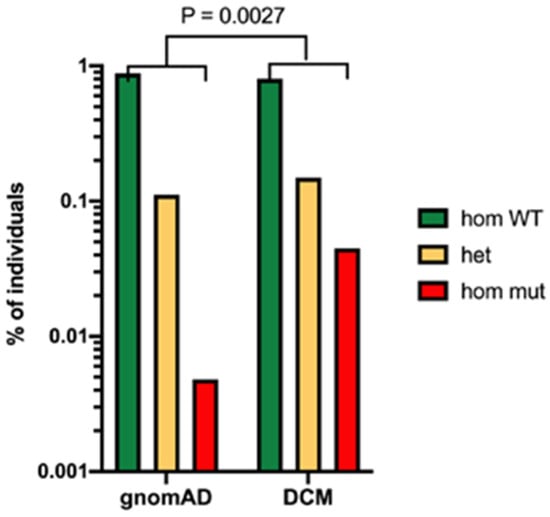
Figure 3. Evidence for the importance of Dectin-1 polymorphism in risk of disseminated coccidioidomycosis. Legend: Frequency of the clec7a mutation in patients with disseminated coccidioidomycosis (DCM) compared to the total population (estimated by gnomeAD). p = 0.0027 Fisher’s exact test.
Mutations in DUOX1 or DUOX1A were also more common in patients with disseminated coccidioidomycosis than controls. DUOX1 and its obligate accessory maturation factor, DUOXA1, are expressed on the apical surface of specific epithelial cells and DUOX1 releases H2O2 in response to calcium-mobilizing agonists [65]. The DUOX mutants did not produce as much H2O2 in response to Dectin-1 agonists as controls [13].
This entry is adapted from the peer-reviewed paper 10.3390/jof10030173
This entry is offline, you can click here to edit this entry!