Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Others
The diagnosis and treatment of patients with mendelian susceptibility to mycobacterial disease (MSMD) pose consistent challenges due to the diverse infection spectrum observed in this population. Common clinical manifestations include Bacillus Calmette-Guérin vaccine (BCG) complications in countries where routine BCG vaccination is practiced, while in non-BCG-vaccinating countries, Non-Tuberculous Mycobacteria (NTM) is prevalent. In tuberculosis-endemic regions, Mycobacterium tuberculosis (MTB) has a high prevalence, along with other intracellular organisms.
- mendelian susceptibility to mycobacterial disease
- non-tuberculous mycobacteria
- mycobacterium tuberculosis complex
1. Introduction
Inborn errors of immunity (IEI) are a group of disorders that result in an impaired immune response [1]. IEIs are classified by the International Union of Immunology Societies (IUIS) depending on the clinical and immunological evaluation of the defects. Mendelian susceptibility to mycobacterial diseases (MSMD) is a rare IEI that is grouped under defects of intrinsic and innate immunity [2,3]. MSMD predisposes to clinical disease caused by weakly virulent mycobacteria such as the Bacillus Calmette-Guérin (BCG) vaccine, nontuberculous mycobacteria (NTM), or environmental mycobacteria (EM). Infections due to more virulent M. tuberculosis have also been reported in MSMD patients. These patients present early in childhood, whereas milder forms can have an onset in adulthood and even remain asymptomatic. Mycobacterial diseases range from localized to disseminated or persistent life-threatening infections. Patients with MSMD may also suffer from infections due to intramacrophagic bacteria, fungi, parasites, and viruses [4,5,6,7,8,9,10,11]. Mutations in 21 different genes (IFNGR1, IFNGR2, IFNG, IL12RB1, IL12RB2, IL23R, IL12B, ISG15, USP18, ZNFX1, TBX21, STAT1, TYK2, IRF8, CYBB, JAK1, RORC, NEMO, SPPL2A, MCTS1, and IRF1) with more than 35 different genetic etiologies of MSMD have been described [12,13,14,15,16,17,18]. These deficiencies exhibit either autosomal recessive, autosomal dominant, or X-linked patterns of inheritance. Confirming the underlying genetic defects in these patients provides a better therapeutic approach and genetic counseling for the affected families. In most suspected MSMD cases, patients are treated empirically without confirming a microbiological and molecular diagnosis.
2. Pathophysiology and Clinical Manifestations of MSMD
Type I cytokines such as IFNγ, IL12, and IL23 play a crucial role in the control of intracellular microorganisms. Once the bacteria are internalized, macrophages recognize the bacterial pathogen-associated molecular patterns and become activated. This activation produces cytokines such as IL12, IL23, and tumor necrosis factor-α (TNF-α). TNF-α plays a role in the formation of granulomas. IL12 leads to the differentiation of T cells into Th1 subpopulations and induces IFNγ secretion. IFNγ further activates the macrophages to eliminate the bacteria. In 1996, the discovery of IFNγR1 deficiency was reported as the first genetic etiology of MSMD with BCG infection. Over the period of discoveries of new genes in MSMD, this condition has been termed an inborn error of IFNγ immunity [7]. To mount an effective immune response against mycobacterial infections, the integrity of the IL12/23/ISG15-IFN-γ circuit is necessary (Figure 1). Mutations in the 21 genes involved in this circuit lead to either an impaired response or the production of IFNγ [8]. Molecular defects in the IL12RB1 and IFNGR1 genes together constitute about 80% of MSMD cases [9].
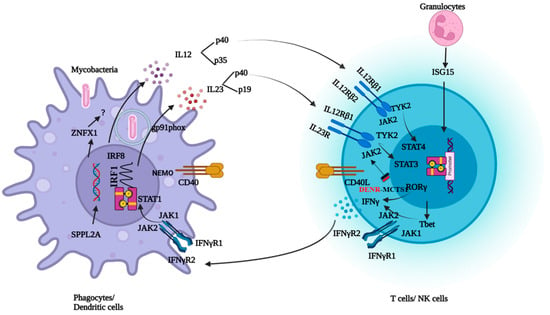
Figure 1. Schematic representation of genes involved in IFNγ response and production.
IL12Rβ1 deficiency is the most common genetic cause of MSMD. IL12 and IL-23 share a common IL12Rβ1 chain encoded by IL12RB1. Therefore, IL12 and IL23 signaling are defective in patients with IL12Rβ1 deficiencies. The clinical phenotype ranges from early deaths in infancy to asymptomatic adulthood, suggesting an incomplete penetrance [19,20]. Patients are vulnerable to mycobacterial disease, Salmonella infections, and chronic mucocutaneous candidiasis (CMC). About 25% of the patients with IL12Rβ1 deficiency develop fungal infections, including CMC [8,9,10]. It is unclear why only some patients with MSMD show susceptibility to fungal infections. Patients with IL-12p40, IL-12Rβ1, or IL-23R deficiency exhibit normal levels of circulating leukocytes from various subsets, including cell types that typically express IL-23R, such as NK, iNKT, mucosal-associated invariant T (MAIT), Vδ2+ γδ T, TH1, TH17, and TH1* cells. However, the susceptibility of certain individuals with deficiencies in IL-12B, IL-12Rβ1, or IL-23R to chronic mucocutaneous candidiasis (CMC) indicates the crucial role of human IL-23 in defending against mucocutaneous Candida spp. infections mediated by IL-17A/F. Recent findings showed that IL-23 induces IL-17A exclusively in MAIT cells from healthy individuals, a response absent in IL-23R-deficient patients. The incomplete penetrance of CMC in these patients is not solely explained by individual variability in IL-17A/F production by leukocyte subsets. Instead, it may be due to the preservation of C. albicans-specific memory CD4+ T cell development in IL-23R-deficient patients and the fact that IL-17A induction is governed by IL-23 in only one lymphocyte subset (MAIT cells), at least in ex vivo conditions [21].
In IL12Rβ1 patients, other clinical manifestations, such as autoimmunity and inflammatory bowel disease (IBD), have been described. Impairment in IL12Rβ1 signaling led to deficiencies in the differentiation of Th17 cells, which are vital for maintaining intestinal homeostasis. Th17 cells contribute to this balance by producing key cytokines, including IL-6, TGF-β, IL-17, and IL-1β, particularly in mucosal damage scenarios. They also play a pivotal role in defending against extracellular pathogens and stimulating the release of antimicrobial peptides from intestinal epithelial cells. The observed reduction in the Th17 cell population in the patient strongly suggests its potential involvement in the development of inflammatory bowel disease in this individual, aligning with the clinical manifestations seen in IL-12Rb1 patients, such as autoimmunity and IBD [22,23,24].
The second most common cause of MSMD is IFNγR1 deficiency. Two forms of autosomal recessive complete IFNγR1 deficiencies are described, with the presence of a non-functional receptor or a receptor that does not express at the cell surface. These complete forms of IFNγR1 are severe forms of MSMD. Patients present early, before 3 years of age, and suffer from disseminated BCG and/or EM with poorly characterized granuloma lesions. Infections with M. tuberculosis, viruses, and bacteria have also been reported [25,26,27,28]. In partial IFNγR1 deficiencies, the receptor is expressed on the cell surface, but the response to IFNγ is impaired. These are less severe forms than a complete IFNγR1 deficiency. In AD with partial IFNγR1 deficiency, there is an accumulation of truncated receptors on the surface due to a mutation in the intracellular domain lacking a receptor recycling motif. Higher amounts of protein are detected on the surface. Multifocal osteomyelitis is commonly seen in these patients. Recently, it was reported that AD IFNγR1-deficient and AD STAT1-deficient cells exhibit resistance to the inhibitory effects of IFNγ on osteoclast differentiation and bone resorption. Additionally, the IFNγ induced inhibition of NFATC1 mRNA, a critical transcription factor involved in osteoclast differentiation, was compromised in both AD IFNγR1-deficient and AD STAT1-deficient cells. These findings suggest that the increased osteoclastogenesis is responsible for the occurrence of osteomyelitis in these patients. Osteomyelitis is relatively rare in patients with complete IFNγR1 and STAT1 deficiency. Both complete AR IFNγR1 deficiency and complete AR STAT1 deficiency are life-threatening conditions, necessitating early intervention through hematologic stem cell transplantation to ensure survival. The severity of these disorders poses challenges for long-term clinical course monitoring, potentially resulting in an underestimation of the prevalence of bone involvement [29,30,31].
IL12p40 deficiency is the third most common deficiency reported in MSMD. Both IL12 and 23 are heterodimers having the IL12p40 subunit in common encoded by IL12B. Both IL12 and IL23 signaling are defective in patients with IL12p40 deficiency. Complete IL12p40 deficiency is a clinical phenocopy of IL12Rβ1 deficiency [20,32].
Unlike IFNγR1 deficiency, two forms of complete IFNγR2 deficiency have been reported with or without cell surface expression. The clinical presentation is similar to that of complete IFNγR1 deficiencies. In partial IFNγR2 deficiency, cell surface expression is weak, and response to IFNγ is impaired. The clinical phenotype is milder. Expression of IFNγR2 is very low in AD IFNγR2 deficiency. These deficiencies display incomplete penetrance [33,34].
Mutations in the STAT1, JAK1, IRF8, SPPL2A, NEMO, and CYBB genes are responsible for phagocytic defects leading to susceptibility to mycobacterial infections. JAK1 is a tyrosine kinase that induces phosphorylation and homodimerization of STAT1 upon binding of IFNγ to the receptor. Impaired response to IFNγ in phagocytes is seen in patients with JAK1 and STAT1 deficiencies. -Various forms of inherited STAT1 deficiency have been documented, with autosomal inheritance causing complete or partial STAT1 deficiency. Complete recessive STAT1 deficiencies are a result of the null mutations. The patient’s cells display unresponsiveness to IFN-γ and IFN-α/β. Consequently, these patients are susceptible to severe infections with mycobacteria and viruses. On the other hand, missense mutations lead to partial recessive STAT1 deficiency, resulting in an impaired but not abolished ability to respond to IFN-γ and IFN-α/β, leading to infections with intracellular bacteria and viruses.
Autosomal dominant STAT1 deficiency can result in loss-of-function (LOF) and gain-of-function (GOF) depending on underlined mutations. LOF mutations alter both IFNγ and IFN-α responses but have a negative dominant effect on the IFNγ response only. These patients typically develop mycobacterial infections without viral infections.
Conversely, mutations resulting in STAT1 gain of function predominantly occurring in the coiled-coil domain and DNA binding domain lead to increased phosphorylation and GAS-binding activity. Cells of affected patients exhibit an enhanced response to IFN-γ, IFN-α, and IL27. These patients commonly develop chronic mucocutaneous candidiasis (CMC) [35,36,37,38,39,40,41,42].
NEMO (NF-kappa-B essential modulator ) is a regulatory subunit of the nuclear factor-ĸB (NFĸB) inhibitor kinase complex. Impaired CD40-dependent IL12 production is responsible for the clinical phenotype of patients with NEMO deficiency [43,44]. TYK2 (Tyrosine kinase 2) is a Janus kinase that, upon stimulation with IL12 and 23, induces phosphorylation and dimerization of STAT4 and STAT3. Mutations in TYK2 lead to susceptibility to mycobacterial and viral infections. Homozygosity for the allele of TYK2 P1104A, is a rare genetic etiology of MSMD that selectively disrupts IL23 responses. Patients with defects in RORC are susceptible to mycobacteriosis and candidiasis due to impaired IFNγ and IL17 immunity [8]. Disease-causing mutations in CYBB are known to cause chronic granulomatous disease. The hemizygous mutations Q231P and T178P in CYBB result in diminished NADPH oxidase activity, specifically in monocyte-derived macrophages. In contrast to individuals with Chronic Granulomatous Disease (CGD), neutrophils, monocytes, and monocyte-derived dendritic cells (MDCs) exhibit an unaffected respiratory burst. Since macrophage respiratory burst is essential in an immune response against mycobacteria, these variants have been included under MSMD [45]. A complete absence of monocytes and dendritic cells is seen in AR IRF8 deficiency, leading to susceptibility to mycobacterial and fungal infections. AD IRF8 deficiency leads to an absence of myeloid dendritic cells and susceptibility to mycobacterial infections. SPPL2A deficiency accumulates CD74 NTF in HLA class II+ myeloid and lymphoid cells. Accumulation of toxic fragments leads to the depletion of IL-12- and IL-23-producing CD1c+ conventional dendritic cells [46]. IL12RB2 and IL23R are the newly discovered but rare deficiencies that also underlie MSMD. Innate lymphoid cells (ILC1 and ILC2) exhibit a response to IL12, while ILC3, on the other hand, demonstrates responsiveness to IL23, resulting in the production of IFNγ. IFNγ production is compensated for in the absence of either of the cytokines [12]. Therefore, IL12RB2 and IL23R defects have lower clinical penetrance than IL12Rβ1 deficiency. In T bet deficiency, patients suffered from BCGosis due to impaired IFNγ production by innate and innate-like lymphocytes [14]. ZNFX1 is essential for monocyte homeostasis, and deficiency leads to susceptibility to mycobacterial disease [15]. Mononuclear myeloid cells from patients with IRF deficiency have an impaired response to IFNγ, leading to susceptibility to mycobacteria [16]. The MAIT and Vδ2+ gδ T-lymphocyte subsets of the MCTS1-deficient patients generate small amounts of IFNγ in response to IL23 and BCG [17]. Impaired immunity due to the monogenic defects in genes involved in the IL12/23/ISG15-IFN-γ circuit results in clinical phenotypes of MSMD.
3. Infectious Spectrum of MSMD
In regions where BCG is not administered at birth, NTM infections are the typical clinical manifestations, while in BCG-administering countries, complications related to BCG are commonly observed. These patients are also vulnerable to intracellular organisms, and the site of infection can range from localized to disseminated, impacting various organs (Figure 2).
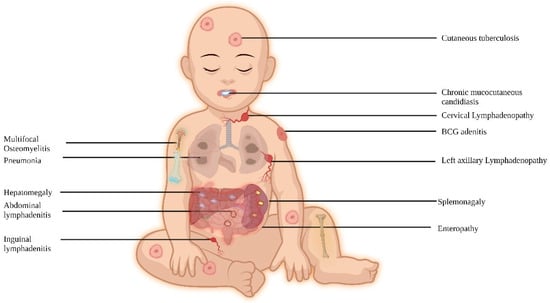
Figure 2. Clinical features of MSMD.
Usually, BCG infection after vaccination is the most common infection in MSMD patients. BCG is a live-attenuated vaccine administered at birth in some countries. It is made from the subculture of Mycobacterial spp. i.e., Mycobacterium bovis (M. bovis). Currently, Brazilian (Moreau/Rio de Janeiro), Danish (Copenhagen–1331), Pasteur-1173, Japanese (Tokyo–172–1), Russian (Moscow–368), and Bulgarian (Sofia–SL222), Connaught-Toronto, Merieux, Chinese-Shanghai, and TICE strains are used for BCG vaccine production around the globe [47]. More than 170 countries give BCG vaccines as part of their national immunization programs. WHO recommends one dose to all neonates in countries with high TB burdens.
In the general population, BCG is proven to be safe, but adverse effects of vaccination can be seen in infants with underlying inborn errors of immunity (IEI) [48,49,50]. BCG-itis is a localized BCG infection observed as a local abscess, a severe ulcer at the injection site, or involvement of the regional ipsilateral ganglia with suppuration, fistula formation, or both. BCGosis is a life-threatening disseminated infection involving distal lymph nodes, skin, lungs, bone, liver, spleen, and the central nervous system (CNS). An adverse effect of the BCG vaccination is usually present within the first 6 months or beyond 12 months. Along with genetic factors, response to BCG vaccination is also dependent on the BCG strain used, storage of the vaccine, inappropriate dose, and injection technique.
This real-world data signifies the screening of MSMD patients in the event of BCG complications from BCG vaccination. Very often, the diagnosis is delayed in patients with BCG complications since they are not investigated. Very often, microbiological evidence is not always available, and a diagnosis is made solely based on clinical findings. This is due to a lack of awareness or diagnostic facilities. Early screening of these patients not only reduces the delay in diagnosis but also reduces morbidity and mortality. BCG vaccination can be delayed in patients with a strong family history of sibling death due to BCG complications and must be investigated.
Very little literature is available on MTB infections in MSMD. A large series of IL12Rβ1 patients reported only 6 patients with isolated Mycobacterium tuberculosis infections [67,68,69,70], and in this group, mycobacterial infections caused by BCG and EM are more common. In countries where BCG vaccination is mandatory, patients develop BCG disease, as a primary course of infection. Once diagnosed with BCG disease these patients do not acquire secondary mycobacterial infections. In TB-endemic countries, BCG disease may prevent subsequent EM disease but not MTB [20]. This is because M. tuberculosis has a close phylogenetic relationship with BCG and is more virulent than EM. Therefore, patients develop recurrent TB infections. The study from South Africa reported 22 patients with MSMD, and 64% of patients were positive for MTBC. Out of which, 55% of patients had recurrent episodes of TB involving the lungs, central nervous system, and gastrointestinal tract [57]. In a cohort of 25 patients diagnosed with IL12Rβ1 deficiency from India, 11 patients developed recurrent TB involving organs such as the lungs, abdomen, brain, skin, and kidney. IL12Rβ1, IFNγR1, STAT1, IL12p40, CYBB, and TYK2 P1104A deficiencies are found to be the commonest deficiencies associated with disseminated, extra-pulmonary, or recurrent TB [9,10,12].
The prevalence of NTM infections is high in countries where BCG vaccination is not administered. NTM mostly affects the lungs, but infections of the skin, soft tissue, and lymph nodes can also be seen [71]. In younger children, single-site lymphadenitis is the most common manifestation of NTM infection in low TB-endemic countries [72]. NTMs are defined as species of the Mycobacterium genus, excluding Mycobacterium tuberculosis (MTB complex) and Mycobacterium leprae. They are also called atypical mycobacteria or environmental mycobacteria since they habitat for food, soil, and water in the environment [73]. To date, 200 species of non-tuberculous mycobacteria have been identified [64]. They are further classified as rapid-growing or slow-growing mycobacteria, depending on how fast they can be grown in the laboratory. NTM diseases due to Mycobacterium avium and the Mycobacterium avium complex are commonly seen in patients with MSMD. These patients present with fever, night sweats, weight loss, abdominal pain, and diarrhea and may present with anemia, hepatosplenomegaly, and lymphadenopathy. Disseminated infection of the lung, spleen, multiple lymph nodes, and vertebrae due to M. colombiense is also reported [54]. Mycobacterium chelonae and Mycobacterium fortuitum are the second-most common infections reported in MSMD patients. Mycobacterium genavense and Mycobacterium simiae cause disseminated NTM disease in MSMD patients. Mycobacterium avium complex comprising M. avium and M. intracellulare, Mycobacterium ulcerans, and Mycobacterium marinum are the slow-growing mycobacteria (SGM). Mycobacterium fortuitum, Mycobacterium abscessus, and Mycobacterium chelonae are rapidly growing mycobacteria (RGM) that cause disseminated NTM infections in MSMD patients. In MSMD patients, 45% of cases with RGM have been reported, which includes Mycobacterium abscessus (32%) and Mycobacterium fortuitum (12%) [71]. High morbidity and up to 52% mortality due to NTM infections have been reported in MSMD patients [53].
This entry is adapted from the peer-reviewed paper 10.3390/pathogens13030203
This entry is offline, you can click here to edit this entry!