Asthma is a chronic inflammatory disease involving reversible airway obstruction, bronchial hyperreactivity (BHR), mucus overproduction, and airway narrowing and remodeling. The symptoms include coughing, wheezing, and breathlessness and may become aggravated at night or during physical activity, resulting in decreased life quality, impaired productivity, and significant utilization of healthcare resources. Younger individuals are affected more frequently than adults: 9.1% of children, 11.0% of adolescents, and 6.6% of adults experience asthma symptoms, and the prevalence across all age groups is increased in high-income countries. The worldwide mortality rate for childhood asthma reaches up to 0.7 deaths per 100,000, and children who suffer from severe asthma have a greater risk of developing chronic obstructive pulmonary disease (COPD) in adulthood. Genes play a larger role in children’s asthma.
- ADAM33
- asthma
- a disintegrin and metalloprotease 33
- airway remodeling
1. ADAM Molecules in General
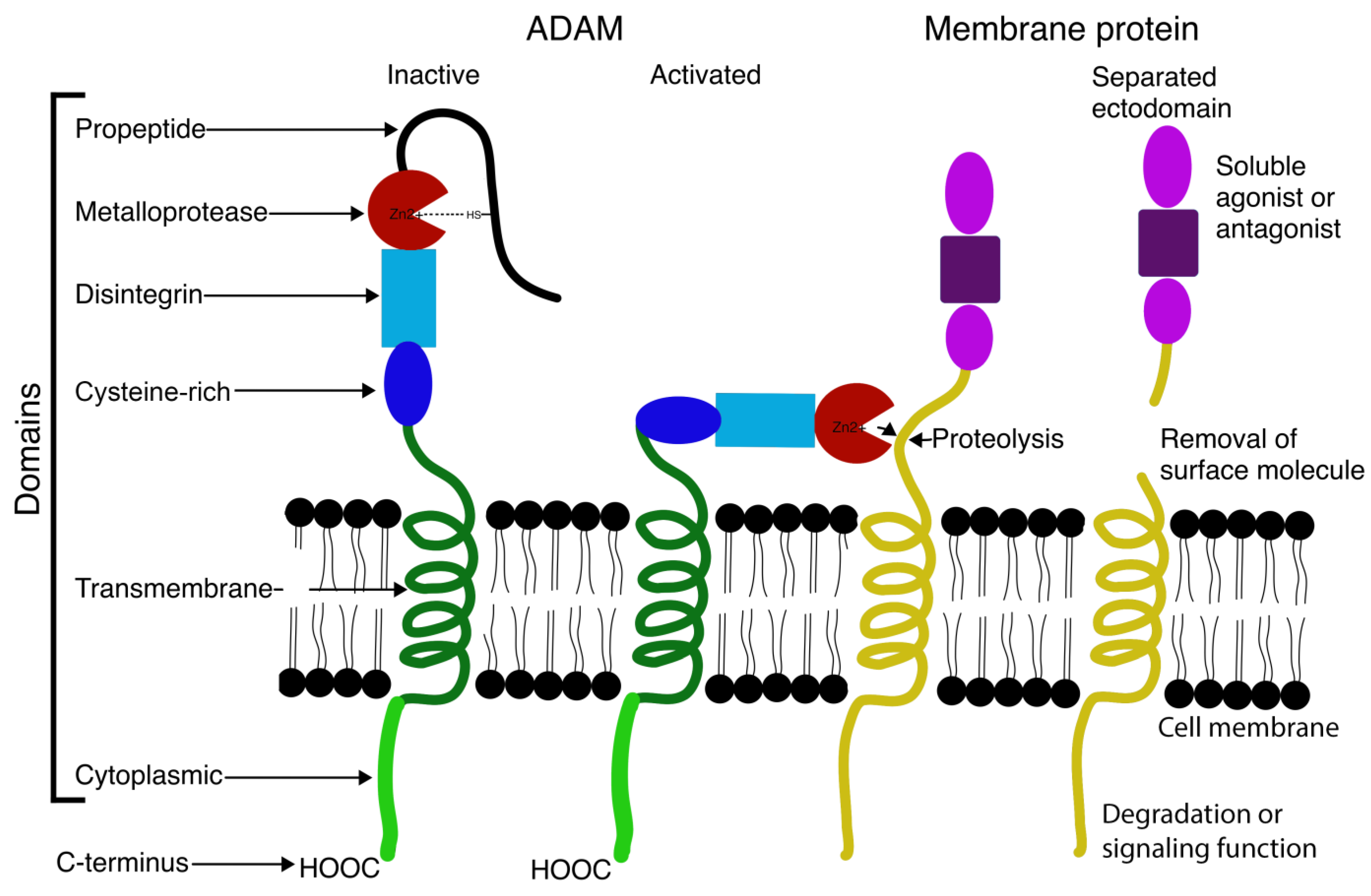
2. Structure
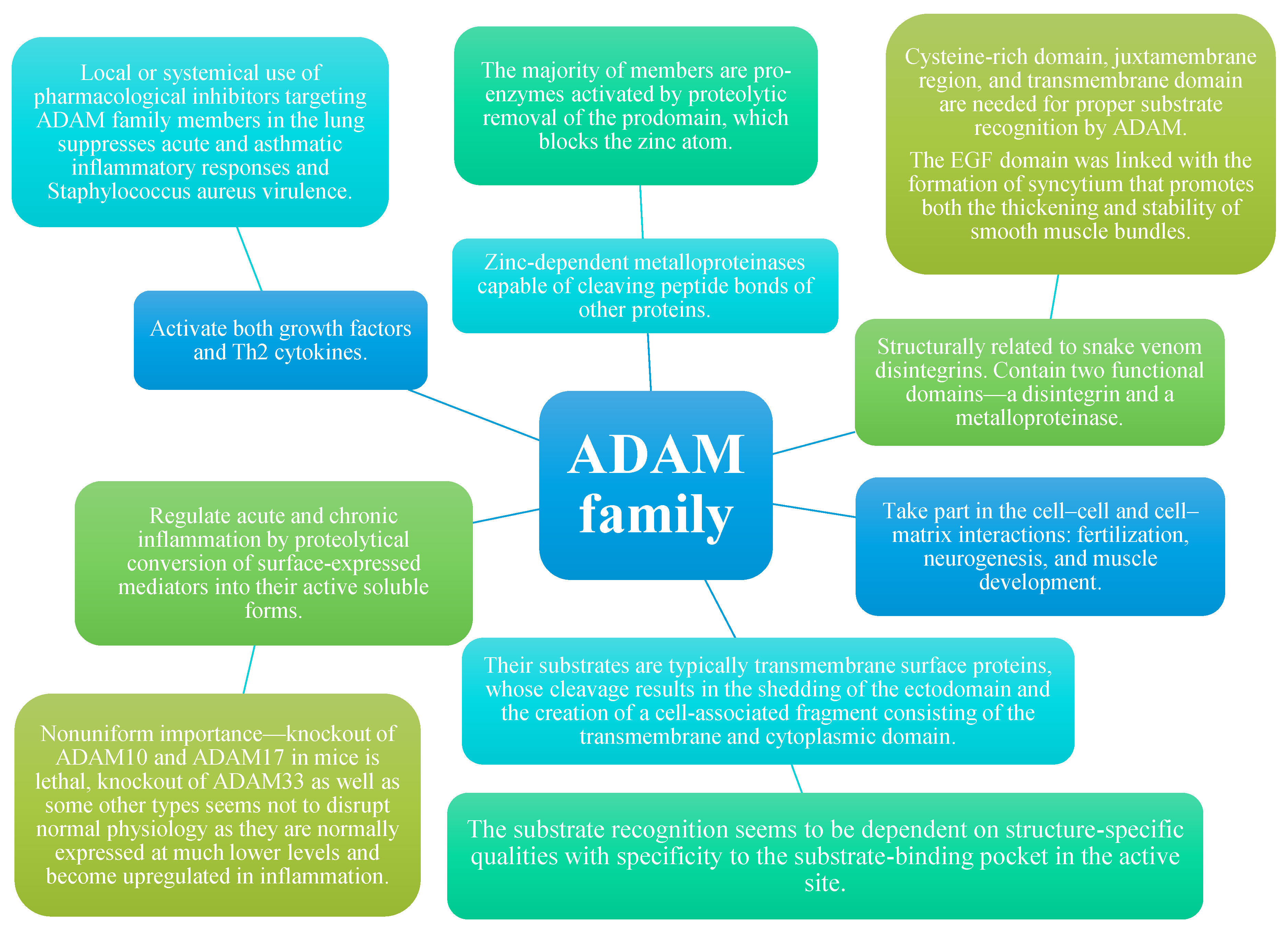
3. Function
This entry is adapted from the peer-reviewed paper 10.3390/ijms25042318
References
- Human Gene ADAM33 (ENST00000356518.7) from GENCODE V43. Available online: https://genome.ucsc.edu/cgi-bin/hgGene?db=hg38&hgg_type=knownGene&hgg_gene=ADAM33 (accessed on 15 February 2023).
- Tripathi, P.; Awasthi, S.; Gao, P. ADAM Metallopeptidase Domain 33 (ADAM33): A Promising Target for Asthma. Mediat. Inflamm. 2014, 2014, 572025.
- Dreymueller, D.; Uhlig, S.; Ludwig, A. ADAM-family metalloproteinases in lung inflammation: Potential therapeutic targets. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L325–L343.
- Holgate, S.T.; Davies, D.E.; Powell, R.M.; Holloway, J.W. ADAM33: A newly identified protease involved in airway remodelling. Pulm. Pharmacol. Ther. 2006, 19, 3–11.
- Li, H.F.; Yan, L.P.; Wang, K.; Li, X.T.; Liu, H.X.; Tan, W. Association between ADAM33 polymorphisms and asthma risk: A systematic review and meta-analysis. Respir. Res. 2019, 20, 38.
- Davies, E.R.; Kelly, J.F.C.; Howarth, P.H.; Wilson, D.I.; Holgate, S.T.; Davies, D.E.; Whitsett, J.A.; Haitchi, H.M. Soluble ADAM33 initiates airway remodeling to promote susceptibility for allergic asthma in early life. JCI Insight 2016, 1, e87632.
- Howard, T.D.; Postma, D.S.; Jongepier, H.; Moore, W.C.; Koppelman, G.H.; Zheng, S.Q.; Xu, J.F.; Bleecker, E.R.; Meyers, D.A. Association of a disintegrin and metalloprotease 33 (ADAM33) gene with asthma in ethnically diverse populations. J. Allergy Clin. Immunol. 2003, 112, 717–722.
- Holgate, S.T.; Davies, D.E.; Rorke, S.; Cakebread, J.; Murphy, G.; Powell, R.M.; Holloway, J.W. ADAM 33 and Its Association with Airway Remodeling and Hyperresponsiveness in Asthma. Clin. Rev. Allergy Immunol. 2004, 27, 023–034.
- Holgate, S.T. Mechanisms of Asthma and Implications for Its Prevention and Treatment: A Personal Journey. Allergy Asthma Immunol. Res. 2013, 5, 343.
- Zheng, W.; Wang, L.; Su, X.; Hu, X.F. Association between V4 polymorphism in the ADAM33 gene and asthma risk: A meta-analysis. Genet. Mol. Res. 2015, 14, 989–999.
- Van Eerdewegh, P.; Little, R.D.; Dupuis, J.; Del Mastro, R.G.; Falls, K.; Simon, J.; Torrey, D.; Pandit, S.; McKenny, J.; Braunschweiger, K.; et al. Association of the ADAM33 gene with asthma and bronchial hyperresponsiveness. Nature 2002, 418, 426–430.
- Powell, R.M.; Wicks, J.; Holloway, J.W.; Holgate, S.T.; Davies, D.E. The Splicing and Fate of ADAM33 Transcripts in Primary Human Airways Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2004, 31, 13–21.
- Shapiro, S.D.; Owen, C.A. ADAM-33 Surfaces as an Asthma Gene. N. Engl. J. Med. 2002, 347, 936–938.
- Hur, G.Y.; Broide, D.H. Genes and Pathways Regulating Decline in Lung Function and Airway Remodeling in Asthma. Allergy Asthma Immunol Res. 2019, 11, 604.
- Baurakiades, E.; Costa, V.H.; Raboni, S.M.; de Almeida, V.R.T.; Larsen, K.S.K.; Kohler, J.N.; Gozzo, P.D.C.; Klassetn, G.; Manica, G.C.; de Noronha, L. The roles of ADAM33, ADAM28, IL-13 and IL-4 in the development of lung injuries in children with lethal non-pandemic acute infectious pneumonia. J. Clin. Virol. 2014, 61, 585–589.
- Yang, Y.; Haitchi, H.M.; Cakebread, J.; Sammut, D.; Harvey, A.; Powell, R.M.; Holloway, J.W.; Howarth, P.; Holgate, S.T.; Dalvies, D.E. Epigenetic mechanisms silence a disintegrin and metalloprotease 33 expression in bronchial epithelial cells. J. Allergy Clin. Immunol. 2008, 121, 1393–1399.e14.
- Moheimani, F.; Hsu, A.C.-Y.; Reid, A.T.; Williams, T.; Kicic, A.; Stick, S.M.; eHansbro, P.M.; Wark, P.A.; Knight, D.A. The genetic and epigenetic landscapes of the epithelium in asthma. Respir Res. 2016, 17, 119.
- Sunadome, H.; Matsumoto, H.; Petrova, G.; Kanemitsu, Y.; Tohda, Y.; Horiguchi, T.; Kita, K.; Kuwabara, K.; Tomii, K.; Fujimura, N.; et al. IL4Rα and ADAM33 as genetic markers in asthma exacerbations and type-2 inflammatory endotype. Clin. Exp. Allergy 2017, 47, 998–1006.
- Andrews, A.; Nasir, T.; Bucchieri, F.; Holloway, J.; Holgate, S.; Davies, D. IL-13 receptor α 2: A regulator of IL-13 and IL-4 signal transduction in primary human fibroblasts. J. Allergy Clin. Immunol. 2006, 118, 858–865.
- Ghaffar, O.; Hamid, Q.; Renzi, P.M.; Allakhverdi, Z.; Molet, S.; Hogg, J.C.; Stephanie, A.S.; Luster, A.D.; Lamkhioued, B. Constitutive and Cytokine-Stimulated Expression of Eotaxin by Human Airway Smooth Muscle Cells. Am. J. Respir. Crit. Care Med. 1999, 159, 1933–1942.
- Jie, Z.; Jin, M.; Cai, Y.; Bai, C.; Shen, Y.; Yuan, Z.; Hu, Y.; Holgate, S. The effects of Th2 cytokines on the expression of ADAM33 in allergen-induced chronic airway inflammation. Respir. Physiol. Neurobiol. 2009, 168, 289–294.
- Song, G.G.; Kim, J.H.; Lee, Y.H. Association between ADAM33 S2 and ST+4 polymorphisms and susceptibility to asthma: A meta-analysis. Gene 2013, 524, 72–78.
- Bora, E.; Arikan-Ayyildiz, Z.; Firinci, F.; Çankaya, T.; Giray-Bozkaya, Ö.; Uzuner, N.; Ülgetnalp, A. ADAM33 Gene Polymorphisms Are Not Associated with Asthma in Turkish Children. Pediatr. Allergy Immunol. Pulmonol. 2012, 25, 97–100.
- Lee, J.Y.; Park, S.W.; Chang, H.K.; Kim, H.Y.; Rhim, T.; Lee, J.H.; Jang, A.S.; Koh, E.S.; Park, C.S. A Disintegrin and Metalloproteinase 33 Protein in Patients with Asthma. Am. J. Respir. Crit. Care Med. 2006, 173, 729–735.
- Foley, S.C.; Mogas, A.K.; Olivenstein, R.; Fiset, P.O.; Chakir, J.; Bourbeau, J.; Pierre, E.; Lemiere, C.; Martin, J.; Hamid, Q. Increased expression of ADAM33 and ADAM8 with disease progression in asthma. J. Allergy Clin. Immunol. 2007, 119, 863–871.
- Tripathi, P.; Awasthi, S.; Husain, N.; Prasad, R.; Mishra, V. Increased expression of ADAM33 protein in asthmatic patients as compared to non-asthmatic controls. Indian J. Med. Res. 2013, 137, 507–514.
- Awasthi, S.; Tripathi, P.; Prasad, R. Environmental risk factors for asthma in Lucknow: A case–control study. Clin. Epidemiol. Glob. Health 2013, 1, 115–123.
- Zhang, Y.; Tan, M.; Qian, X.; Li, C.; Yue, L.; Liu, Y.; Shi, S. Interaction between early-life pet exposure and methylation pattern of ADAM33 on allergic rhinitis among children aged 3–6 years in China. Allergy Asthma Clin. Immunol. 2021, 17, 44.