Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Life-threatening systemic fungal infections occur in immunocompromised patients at an alarming rate. Current antifungal therapies face challenges like drug resistance and patient toxicity, emphasizing the need for new treatments. Membrane-bound enzymes account for a large proportion of antifungal targets, especially ones that contribute to cell wall and cell membrane biosynthesis. Moreover, structural biology has led to a better understanding of the mechanisms by which these enzymes synthesize their products, as well as the mechanism of action for some antifungals.
- cryo-EM
- structure biology
- membrane-bound enzymes
- antifungal development
- drug resistance
- rational drug design
1. Introduction
Over the past three decades, fungal infections in humans have surged due to a rise in immunocompromised patients [1,2,3]. The most common culprits behind these infections are Candida species, with Candida albicans being the most prevalent. C. albicans is known for causing a wide range of infections, including those affecting the mucous membranes, skin, and bloodstream [2,4,5]. Effective treatment requires antifungal therapy, with the main classes being azoles, echinocandins, and polyenes. Each antifungal class targets specific fungal cellular components, but they face challenges like growing drug resistance and patient toxicity [6,7,8,9,10]. Hence, there is a pressing need for new antifungals.
In a quest to address this challenge, various strategies have been exploited to develop new drugs. High-throughput screening (HTS) and rational drug design are two ways to identify leads for drug development [11,12]. For HTS, target-based and whole-cell-based screenings are two major approaches, and both require investigators to screen thousands of compounds, which requires enormous physical resources, both chemically and mechanically [13]. On the contrary, rational drug design develops drugs based on information from the structure, function, and mechanism of action of the target protein and can also aid in optimizing hits identified from HTS [11]. Rational drug design also comes in two types: ligand-based and structure-based approaches. The former depends on understanding the structure of existing ligands that can bind to a target, while the latter focuses on designing inhibitors using the structural details of target proteins [14]. The rational drug design process typically involves multiple rounds of design, synthesis, and evaluation to yield compounds potent and specific enough for preclinical trials [15]. The atomic structures, as well as predicted structures from various methods, of the target proteins have been shown to be useful in drug design [16,17]. Dorzolamide, used for treating glaucoma [15], and saquinavir, an HIV protease inhibitor [18], are two market drugs refined or conceived through the structure-based method.
In recent years, minimal progress has been made to identify new antifungals because of the relatively high gene homology (~40%) and conservation of fundamental biochemical pathways between fungi and humans. This similarity prohibits easy identification of drugs that are selectively toxic to fungi [19]. To this end, it is important to identify fungal-specific targets that are either absent in mammals, or can be inhibited specifically enough not to cause side effects in mammals, and can thus provide selective inhibition of fungi. The most effective of these molecules in current use all target the cell envelope (cell wall–cell membrane complex). These include the antifungal classes of echinocandins, azoles, allylamines, and polyenes, which act on fungal cell walls, cell membrane biosynthesis, and membrane integrity, respectively. There are other inhibitors known to target non-envelope proteins as well, but they are less commonly used in treating invasive infections in humans. They target nucleic acid biosynthesis, the respiratory chain, and microtubule function. For example, pyrimidine analogs, such as flucytosine, inhibit fungal thymidylate synthase, affecting DNA and RNA synthesis [20]. However, the major drawbacks of flucytosine include widespread occurrence of resistance in many fungal species and bone marrow toxicity in patients [21,22,23,24]. Benzimidazoles, such as thiabendazole, disrupt microtubule function, leading to an inhibition of fungal cell division and, ultimately, cell death [25]. Thiabendazole has been used to treat a variety of plant fungal infections, and to a lesser extent, to treat fungal infection in animals and humans because of its narrow spectrum of activity and the potential for liver toxicity [26,27]. Similarly, succinate dehydrogenase inhibitors (SDHIs) and quinone outside inhibitors, targeting succinate dehydrogenase and cytochrome bc1 complex (complex III), respectively, disrupt energy production in fungi and have been used in agriculture to combat molds and fungi [28,29]. In addition, novel agents like olorofim (targeting dihydroorotate dehydrogenase) and fosmanogepix (targeting inositol acyltransferase) possess broad spectrum activity and remarkable novelty that are expected to be significant in the future [30,31,32,33,34].
2. Cell Wall Biosynthesis Enzymes
The fungal cell wall is an ideal target for antifungal drugs as it is an organelle that is not conserved in mammals. The cell wall shields fungi from environmental threats and prevents harmful macromolecules from entering the cell [35]. The fungal cell wall accounts for around 40% of the entire cell volume and is made of polysaccharides (mainly glucan and chitin) and glycoproteins [36]. Structurally, chitin and β-1,3-glucan are essential constituents of most fungal cell walls, and they create a gel-like matrix that in some fungi can include α-1,3-glucans, β-1,6-glucans, and glycoproteins. The synthesis of chitin and glucan is mediated by membrane-bound chitin synthases and glucan synthases, respectively, which are effective targets for antifungal drugs. Table 1 lists the antifungal drugs or inhibitors on the market or in the developmental stage that target membrane-bound enzymes involved in cell wall biosynthesis.
Table 1. Antifungal drugs or inhibitors targeting membrane-bound enzymes in cell wall biosynthesis.
| Drug Class/Agent | Structure of an Exemplar Compound | Target Enzyme | Mechanism of Action | Discovery Stage | Is the Drug–Target Interaction Known? | Reference |
|---|---|---|---|---|---|---|
| Polyoxins | 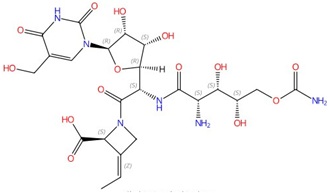 Polyoxin A |
Chitin synthase | Inhibit chitin synthesis in cell wall | Research and development | Yes | [37,38,39] |
| Nikkomycins | 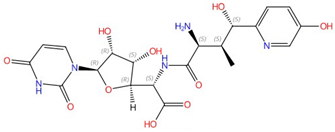 Nikkomycin Z |
Chitin synthase | Inhibit chitin synthesis in cell wall | Clinical trials | Yes | [39,40,41,42] |
| Arthrichitin | 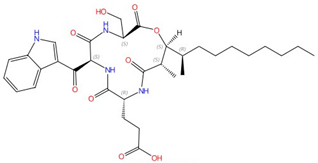 |
Chitin synthase | Inhibits chitin synthesis in cell wall | Research and development | No | [43] |
| Radicicol | 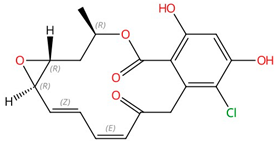 |
Chitin synthase | Inhibits chitin synthesis in cell wall | Research and development | No | [44] |
| Echinocandins (caspofungin, micafungin, anidulafungin and rezafungin) | 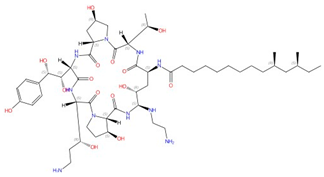 Caspofungin |
β-1,3-glucan synthase | Inhibit cell wall glucan synthesis | Approved | Yes | [45,46,47,48,49] |
| Ibrexafungerp | 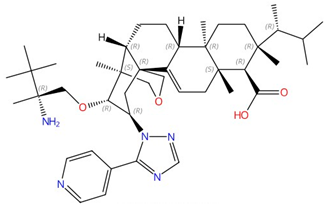 |
β-1,3-glucan synthase | Inhibits cell wall glucan synthesis | Approved for treating vulvovaginal candidiasis | No | [50,51] |
| Pneumocandin A-E | 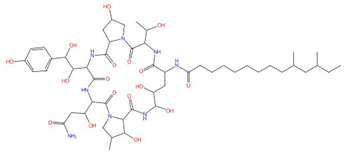 Pneumocandin A0 |
β-1,3-glucan synthase | Inhibits cell wall glucan synthesis | Research and development | No | [52,53,54] |
| Aculeacin A-G | 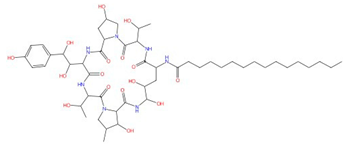 Aculeacin A |
β-1,3-glucan synthase | Inhibits cell wall glucan synthesis | Research and development | No | [55,56] |
| Mulundocandin |  |
β-1,3-glucan synthase | Inhibits cell wall glucan synthesis | Research and development | No | [57,58] |
| Enfumafungin |  |
β-1,3-glucan synthase | Inhibits cell wall glucan synthesis | Research and development | No | [59] |
| Arundifungin |  |
β-1,3-glucan synthase | Inhibits cell wall glucan synthesis | Research and development | No | [60] |
| Papulacandins |  |
β-1,3-glucan synthase | Inhibit cell wall glucan synthesis | Research and development | No | [61] |
3. Cell Membrane Biosynthesis Enzymes
Drugs that impact cell membrane integrity have seen significant success as well [87]. For example, drugs targeting ergosterol synthesis, such as imidazoles and triazoles, are especially effective. While many new antifungal triazole compounds have been introduced recently, they still have a long journey before being recognized as successful antifungals [88]. Beyond sterols, the membrane also contains essential components like phospholipids and sphingolipids, which are crucial for cellular operations and signaling pathways. Table 2 lists antifungal drugs or inhibitors on the market or in developmental stages that target membrane-bound enzymes involved in cell membrane biosynthesis.
3.1. Ergosterol Biosynthesis Enzymes
3.1.1. Lanosterol 14α-Demethylase (Erg11)
Ergosterol is a major component of fungal cell membranes and plays a role similar to cholesterol in human cell membranes [89,90,91]. Its biosynthesis pathway in fungi is a prime target for antifungal drug development, and a detailed ergosterol biosynthesis pathway including all involved enzymes and the sites of inhibition are summarized in [89]. These enzymes, residing in the endoplasmic reticulum and other organelles, are crucial for the synthesis of ergosterol, the main sterol in fungal cell membranes. For example, lanosterol 14α-demethylase (known as Erg11 or CYP51) is a single-pass membrane-bound cytochrome P450 enzyme and is a well-studied drug target of azoles [92,93]. Erg11 is responsible for the demethylation of lanosterol, a step vital for the subsequent conversion of lanosterol to ergosterol, inhibition of which leads to the accumulation of toxic intermediates, thus compromising cell membrane integrity [93]. The structures and their interactions with different azoles were identified using X-ray crystallography for Saccharomyces cerevisiae, Aspergillus fumigatus, Candida albicans, and Candida glabrata Erg11 [92,94,95,96]. Like other members within the P450 enzyme family, Erg11 has a thiolate–heme iron center, the active site where oxidation reactions occur. Also, it contains hydrophobic pockets and channels that accommodate the lipid substrate and facilitate its access to the catalytic center [92,94,95,96]. The interaction between itraconazole and C. albicans Erg11 shows that on a molecular scale, one of the nitrogen atoms in the azole ring binds as the sixth coordinating ligand to the heme iron, preventing oxygen activation [97]. The interaction of the azole ring with the heme is crucial in determining the binding of azole drugs to Erg11 targets in other structures as well [94,95,96]. With the detailed structural information of Erg11 available, structure-directed drug discovery can be performed, which involves virtually screening compound libraries for molecules that might bind more effectively to Erg11, even in resistant enzymes, or designing new molecules based on insights from the enzyme’s structure [97].
Table 2. Antifungal drugs or inhibitors targeting membrane-bound enzymes in cell membrane biosynthesis.
| Drug Class/Agent | Structure of an Exemplar Compound | Target Enzyme | Mechanism of Action | Discovery Stage | Is the Atomic Structure Solved for the Target? | Is the Drug–Target Interaction Known? | Reference |
|---|---|---|---|---|---|---|---|
| Azoles (e.g., fluconazole, itraconazole) | 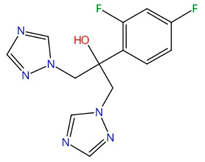 Fluconazole |
Lanosterol 14α-demethylase (Erg11) | Inhibit ergosterol biosynthesis | Approved | Yes | Yes | [92,94,95,96] |
| Allylamines (e.g., terbinafine) | 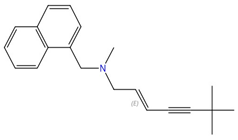 Terbinafine |
Squalene epoxidase (Erg1) | Inhibit ergosterol biosynthesis | Approved for treating topical and oral fungal infections | Yes | Yes | [98,99,100,101] |
| Tomatidine |  |
C-24 sterol methyltransferase (Erg6) | Inhibits ergosterol biosynthesis | Research and development | No | No | [102] |
| Arylguanidines (e.g., abafungin) | 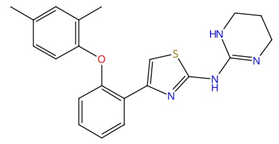 Abafungin |
C-24 sterol methyltransferase (Erg6) | Inhibit ergosterol biosynthesis | Research and development | No | No | [103] |
| H55 |  |
C-24 sterol methyltransferase (Erg6) | Inhibits ergosterol biosynthesis | Research and development | No | No | [104] |
| Morpholines (fenpropimorph, fenpropidin, amorolfine, and Sila-analogue 24) |  Fenpropimorph |
Sterol C-14 reductase (Erg24) and sterol C-8,7 isomerase (Erg2) | Inhibit ergosterol biosynthesis | Research and development | No | No | [105] |
| Sphingofungins |  Sphingofungin A |
Serine palmitoyltransferase (SPT) | Inhibit sphingolipid biosynthesis | Research and development | No | No | [106,107] |
| Lipoxamycin |  |
Serine palmitoyltransferase (SPT) | Inhibits sphingolipid biosynthesis | Research and development | No | No | [108,109] |
| Fumonisins (e.g., fumonisin B1) | 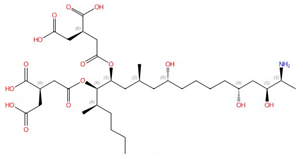 Fumonisin B1 |
Ceramide synthase | Inhibit sphingolipid biosynthesis | Research and development | No | No, but a model was proposed in [110] | [110,111,112] |
| Rustmicin | 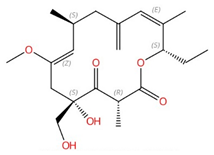 |
Inositol phosphorylceramide (IPC) synthase | Inhibits sphingolipid biosynthesis | Research and development | No | No | [113,114] |
| Khafrefungin | 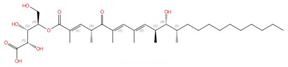 |
Inositol phosphorylceramide (IPC) synthase | Inhibits sphingolipid biosynthesis | Research and development | No | No | [115] |
| Aureobasidin A | 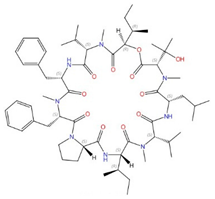 |
Inositol phosphorylceramide (IPC) synthase | Inhibits sphingolipid biosynthesis | Research and development | No | No | [116,117] |
| Haplofungin | 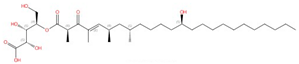 |
Inositol phosphorylceramide (IPC) synthase | Inhibits sphingolipid biosynthesis | Research and development | No | No | [118,119] |
| YU253467 and YU254403 |  YU254403 |
Phosphatidylserine decarboxylase | Inhibit phospholipid biosynthesis | Research and development | No | No | [120] |
| CBR-5884 |  |
Phosphatidylserine synthase | Inhibits phospholipid biosynthesis | Research and development | No | No | [121] |
3.1.2. Squalene Synthase (Erg9)
Besides Erg11, other membrane-bound enzymes are also involved in ergosterol biosynthesis. For example, squalene synthase (Erg9), an enzyme that catalyzes the conversion of two molecules of farnesyl pyrophosphate to squalene, is also a potential drug target due to the functional difference between animal and fungal counterparts [122,123,124]. The structures of Erg9 homologs from a trypanosomatid parasite, Trypanosoma cruzi, and humans were first determined via X-ray crystallography; this could aid in the development of anti-Chagas disease and cholesterol-lowering drugs [125,126,127]. Zaragozic acids are potent competitive inhibitors of rat liver squalene synthases and can potentially treat hypercholesterolemia [128]. Malwal et al. reported the first fungal Erg9 structure from Aspergillus flavus in both apo- and substrate-bound forms and compared it to previous structures [122]. The transmembrane domains of A. flavus Erg9 have similar architectures compared to their human counterparts, but the B-helix is significantly shorter in human Erg9 [122]. This difference might lead to different ligand/inhibitor binding between human and pathogen proteins and could be used in antifungal design.
3.1.3. Squalene Epoxidase (Erg1)
Following the formation of squalene, squalene epoxidase (also known as Erg1) adds an epoxide group to squalene to form 2,3-oxidosqualene [89,122]. Erg1 is also a membrane-bound enzyme located in the ER membrane and its reaction is the rate-limiting step of ergosterol biosynthesis in fungi and cholesterol biosynthesis in mammals. It is predicted to form a complex with Erg9 in the microsomal fraction [129,130]. Terbinafine, an allylamine drug (Figure 6A, Table 2), inhibits Erg1 and leads to ergosterol depletion and accumulation of squalene, which is fungicidal for filamentous fungi but fungistatic for most Candida species [98,99,100]. The human Erg1 structure was solved with an N-terminally truncated enzyme (118–574) in the presence of a known inhibitor NB-598 [101]. The terbinafine molecule was superposed with NB-598 to show the potential mode of inhibition of the molecule (Figure 6B). The conserved residues of human Erg1, L326, L473, F477, F492, F495, L508, P505, and H522 are predicted to form non-polar interactions with the inhibitor, and the mutations of these equivalent residues lead to terbinafine resistance in fungi [101,131,132,133]. Furthermore, a homology model of S. cerevisiae Erg1 was made and compared to its human counterpart [129]. S. cerevisiae Erg1 possesses an extended loop between β-strands 6 and 7 (residues 109–139, based on S. cerevisiae, pointed by the arrow), while the human homolog has a much shorter loop (residues 210–220) (Figure 6C). This compact loop in the human version might aid in crystal formation and the extended loop in S. cerevisiae Erg1 might obstruct this process [129]. Currently, the function of the extended loop in S. cerevisiae Erg1 is unknown, but may be targeted to develop molecules that destabilize the protein or disrupt potential protein–protein interactions.
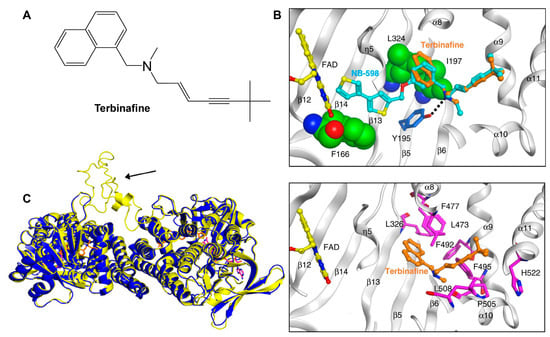
Figure 6. Binding model of terbinafine to squalene epoxidase. (A) Structure of terbinafine. (B) The upper panel shows a superposition of terbinafine (orange) with NB-598 (cyan) in human squalene epoxidase (PDBID:6C6P). The lower panel shows the positions of the known terbinafine-resistant mutations (pink) with respect to terbinafine (orange) in human squalene epoxidase with a superposed terbinafine model. (C) Superposition of human squalene epoxidase structure (blue) and S. cerevisiae squalene epoxidase homology model (yellow). The extended loop on S. cerevisiae Erg1 is pointed to by an arrow. Figure (B) was originally generated in [101] (under Creative Commons CC BY license), and Figure (C) was originally generated in [129] (under Creative Commons CC BY license).
3.1.4. C-24 Sterol Methyltransferase (Erg6)
C-24 sterol methyltransferase (known as Erg6) is also a membrane enzyme involved in ergosterol biosynthesis which was suggested to be an antifungal target due to its absence in mammals [89,134]. Erg6 catalyzes the methylation of the 24th carbon in the sterol side chain in the later stages of ergosterol biosynthesis, the disruption of which leads to reduced mating capability, diminished tryptophan uptake, increased permeability, and susceptibility to cations and antifungals in S. cerevisiae [135,136,137]. In C. albicans, the disruption of Erg6 leads to increased sensitivity to cycloheximide, terbinafine, fenpropimorph, and tridemorph, but not to azoles, while showing resistance to amphotericin B [138]. Interestingly, deletion of Erg6 leads to reduced virulence but not cell growth [139].
Several sterol analogs were determined to suppress Erg6 activity due to their structural resemblance to the substrate or product of Erg6 [140]. Other inhibitors of Erg6, such as tomatidine and arylguanidines (Table 2), effectively hinder the growth of C. albicans, but might inhibit additional cellular targets since disruption of Erg6 does not lead to growth defects [102,103,139,141]. Recently, an antipyrine derivative, H55 (Table 2), identified from screening, showed low cytotoxicity and effectively inhibited C. albicans hyphal formation under various conditions, and also exhibited therapeutic efficacy in mouse models of azole-resistant candidiasis [104]. Various assays support the hypothesis that H55 is an allosteric inhibitor for Erg6, and a molecular dynamics simulation predicts that H55 competes with S-adenosylmethionine for binding to Erg6 [104]. More structural information is needed to validate or provide more insight into the interaction between Erg6 and H55.
There are currently no structures experimentally solved for Erg6 to our knowledge, but Azam et al. modeled a C-24 sterol methyltransferase from Leishmania infantum and identified relevant residues that interact with itraconazole and amphotericin B [134]. Since the substrate-binding sites and active sites are conserved between L. infantum and S. cerevisiae C-24 sterol methyltransferase, ligand/protein interaction information from the L. infantum Erg6 homolog can potentially be applied in antifungal design [134].
3.1.5. Sterol C-14 Reductase (Erg24) and Sterol C-8,7 Isomerase (Erg2)
Morpholines (Table 2) are known to inhibit sterol C-14 reductase (known as Erg24) and sterol C-8,7 isomerase (Erg2), which are two membrane-bound enzymes involved in ergosterol biosynthesis [89,142,143]. Morpholines such as fenpropimorph, fenpropidin, and amorolfine, as well as a silicon containing analog named Sila-analogue 24, exhibit potent antifungal effects against different human fungal pathogens [105]. An erg24∆∆ mutant has reduced virulence in a mouse model of disseminated candidiasis [144]. Erg24 catalyzes the reduction of the C14=15 double bond of sterol intermediates, so sterol intermediates that are not processed by Erg24 cannot be recognized by downstream enzymes, thus perturbing the membrane [89]. Erg2 facilitates the formation of a double bond from the 8 to the 7 position in the sterol intermediate fecosterol, and this enzyme has a polyvalent high-affinity drug binding site similar to that in mammalian sigma receptors [145]. Currently, neither the structures of Erg24 and Erg2 nor their interactions with morpholines have been characterized. The human Erg2 homolog has a solved structure, and its interaction with the anti-breast cancer drug tamoxifen and the cholesterol biosynthesis inhibitor U18666A have been studied [146].
Another sterol C-14 reductase is Erg23, and one bacterial homolog, Methylomicrobium alcaliphilum sterol C-14 reductase, show an interesting arrangement of ten transmembrane domains, with the catalytic domain localized in the carboxy-terminal half (TM6–10). This domain surrounds two linked pockets, with one facing the cytoplasm, which accommodates NADPH, and the second pocket accessible from the lipid bilayer [147]. However, neither the structure of human sterol C-14 reductase nor Methylomicrobium alcaliphilum sterol C-14 reductase have direct use in the antifungal design, and efforts are needed in structure determination of their fungal counterparts.
3.2. Sphingolipid Biosynthesis Enzymes
Sphingolipid production is crucial for the growth and survival of various human fungal pathogens, such as Histoplasma capsulatum and C. albicans [148,149]. Therefore, using a drug to obstruct this process could effectively halt their growth and trigger cell death. The sphingolipid biosynthesis pathway in S. cerevisiae is depicted in Figure 7 from the first step to the formation of the major sphingolipid mannose-(inositol-P)2-ceramide (M(IP)2C) [117,150,151]. Three membrane-bound enzymes involved in sphingolipid synthesis have been suggested as potential antifungal targets—serine palmitoyltransferase (SPT), ceramide synthase, and inositol phosphorylceramide (IPC) synthase. These all have their respective inhibitors.
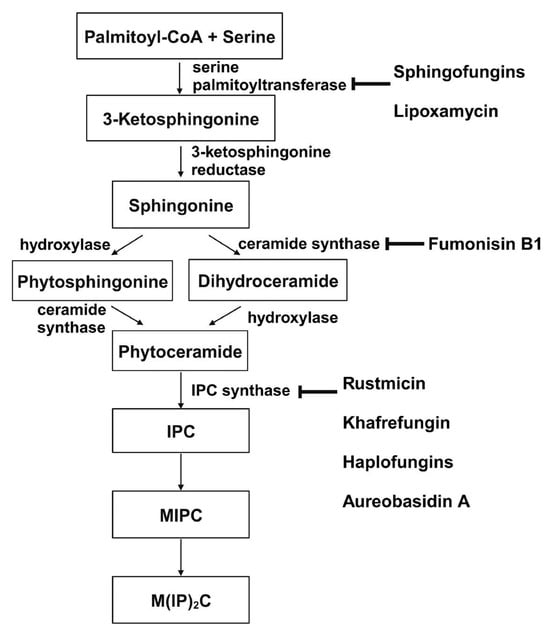
Figure 7. De novo synthesis of sphingolipids in S. cerevisiae and known inhibitors targeting each enzyme. This figure is adapted and modified from [91] (under Creative Commons CC BY license). IPC: Inositolphosphoryl-ceramide; MIPC: mannose inositol-P-ceramide; M(IP)2C: mannose-(inositol-P)2-ceramide.
3.2.1. Serine Palmitoyltransferase (SPT)
SPT catalyzes the condensation of serine and palmitoyl-CoA, which is the first and rate-limiting enzyme in the biosynthesis of sphingolipids [152,153]. SPT uses pyridoxal phosphate (PLP) as a cofactor for catalysis, and belongs to the allene oxide synthase (AOS) family [154]. The active yeast SPT enzyme is a heterodimer made from subunits encoded by lcb1 or lcb2 [155,156], and a third subunit, Tsc3p, is required for high-level SPT activity [157]. The protein structure of the SPT complex was first solved in bacteria [158,159,160], but since the bacterial SPT homologs are soluble homodimers, they provided limited insights into the catalytic mechanism of eukaryotic SPT. In 2021, structures of the human SPT–ORMDL3 complex (ORMDL proteins function as regulatory subunits) in different catalytic states were solved [161,162], and later, the ceramide-sensing mechanism of the SPT-ORMDL3 complex was studied from a ceramide-bound structure [163]. Due to the high sequence similarity between fungal and mammalian SPT subunits [164], the human SPT-ORMDL3 complex can be used to study the mechanism of SPT inhibitors such as sphingofungins [106,107] and lipoxamycin [108,109] (Table 2).
3.2.2. Ceramide Synthase
Ceramide synthase, another membrane-bound enzyme involved in sphingolipid biosynthesis, adds a fatty acyl chain from fatty acyl–coenzyme A (CoA) to the sphingoid base sphinganine to form ceramide [117]. Mammals possess six ceramide synthase isoforms that differ in their tissue distribution and substrate specificity, and each isoform is known to produce ceramides with different acyl chain lengths [165,166]. Currently, there is no structure available for any fungal ceramide synthase homologs to our knowledge, but some studies have provided structure-function characterization of ceramide synthases. Ceramide synthase belongs to the longevity assurance gene 1 (Lag1) protein family, which has a stretch of 52 amino acids that form a highly conserved Lag1p motif [167]. Two conserved histidine residues within this Lag1p motif are crucial for the catalysis and binding of the substrates, the alteration of which negatively impacts the enzymatic function of mammalian and yeast ceramide synthases [167,168,169]. Fumonisins are a group of mycotoxins that have a striking structural resemblance to sphingolipids and are carcinogenic [111] (Table 2). Notably, fumonisin B1 (FB1), among many fumonisins, effectively inhibits ceramide synthase, conferring toxicity and carcinogenic properties. Although neither the protein structure of ceramide synthase nor the FB1/protein binding is solved, an inhibition model for FB1 was proposed [110]. Briefly, concentrations of both substrates affected the potency of FB1, suggesting that FB1 is a competitive inhibitor that binds to the active site of ceramide synthase [112]. It was later found that the tricarballylic acid sidechains play essential roles in the inhibition of FB1, as eliminating tricarballylic acid sidechains reduces the strength of ceramide synthase inhibition in vitro by 10-fold [110,170]. Moreover, this model was further supported by the observation that the FB1 derivative without tricarballylic acid sidechains can be used as a substrate by ceramide synthase, indicating those side chains are required for inhibition [171]. Further structure investigation is needed to validate this model and will also help optimize FB1 to act specifically against fungi.
3.2.3. Inositol Phosphorylceramide (IPC) Synthase
Unlike serine palmitoyltransferase and ceramide synthase, which have homologs in mammalian cells, inositol phosphorylceramide (IPC) synthase catalyzes a reaction unique to plants and some microbial eukaryotes, such as fungi and kinetoplastids. This reaction is the transfer of phosphoinositol from phosphatidylinositol to phytoceramide [117]. Following its discovery in S. cerevisiae, IPC synthases have been characterized in plants [172,173,174] and various protozoans causing neglected tropical diseases, such as Chagas disease and leishmaniasis [175,176,177,178,179]. An alignment of trypanosomatid IPC synthases showed conserved arginine, histidine, and aspartate residues in the active site, and their contributions to a predicted catalytic transfer of the phosphoryl group were demonstrated in Leishmania major IPC synthase [179].
Four inhibitors that act specifically against IPC synthases have been identified (Table 2). Rustmicin, a 14-membered macrolide, is especially potent against C. neoformans, where it inhibits the growth of C. neoformans and its sphingolipid synthesis at concentrations <1 ng/mL, with an IC50 of 70 pM against solubilized C. neoformans IPC synthase [113,114]. The compound khafrefungin, isolated from a Costa Rican plant, displayed antifungal effects again C. albicans and C. neoformans and was determined to inhibit C. albicans IPC synthase with an IC50 of 0.6 nM, but with no effects on mammalian sphingolipids [115]. Another compound, aureobasidin A, a natural compound from the fungus Aureobasidium pullulans, has very low (sub-μg/mL) MIC values for S. cerevisiae, C. albicans, and C. neoformans with IC50 values for IPC synthase activities ranging from 0.2 to 4.9 nM [116,117]. Haplofungin, a phytoceramide mimic isolated from the fungus Lauriomyces bellulus, also showed potent inhibitory activities against fungal IPC synthases [118,119]. However, despite the fact that several potent IPC synthase inhibitors have been identified, the atomic structure of this protein is unsolved. The Arabidopsis thaliana IPC synthase monomer is predicted to have six transmembrane domains with a flexible N-terminal region (AlphaFoldDB: Q9SH93), and further structure–activity relationship studies will be helpful for optimizing current inhibitors or designing new antifungal drugs.
3.3. Phospholipid Biosynthesis Enzymes
Phospholipids, accounting for 40–60% of lipids in eukaryotic cells, are the predominant lipids present in most organisms’ membranes [180]. The four major phospholipids in eukaryotes are phosphatidylserine (PS), phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylinositol (PI) [181]. PC and PE constitute the majority of phospholipids in yeasts and are required for functional membrane construction in eukaryotic organisms. They are involved in membrane integrity and mitochondrial functions [182,183]; PI species are involved in various cellular signal transduction pathways [184]. PS is enriched in the plasma membrane [181] and is involved in a variety of other signaling cascades such as the activation of protein kinase C [185,186,187]. PS is required for virulence in C. albicans and viability in C. neformans [183,188,189], but also plays important roles in apoptosis and blood clotting in mammals [190,191,192]
Phospholipid biosynthesis in cells is intricate, with distinct variations between fungal and mammalian cells. The phospholipid biosynthesis pathways of C. albicans (A) and mammals/parasites (B) are shown in Figure 8, adapted from [193]. The biosynthesis of phospholipids in C. albicans differs from mammalian cells in several steps. First, mammalian cells encode one PSD gene for PS decarboxylase [194], which converts PS to PE, while yeast has two distinct proteins with little similarity, PSD1 and PSD2. Each of these genes has PS decarboxylase activity [195,196]. Also, the production of PS uses a different mechanism in mammalian versus fungal cells. In mammalian cells, PS is produced through a base-exchange reaction catalyzed by the mammalian phosphatidylserine synthase-1 (PSS1) and phosphatidylserine synthase-2 (PSS2), in which the headgroups of existing PC and PE, respectively, are cleaved off and replaced with serine to produce PS [197]. On the contrary, fungal cells condense cytidine diphosphate diacylglycerol (CDP-DAG) and serine into PS via phosphatidylserine synthase (PS synthase), using a different catalytic mechanism compared to the mammalian PSS1/PSS2 enzymes [181,197]. This section will discuss the current inhibitors and structure studies of PS decarboxylase and PS synthase.
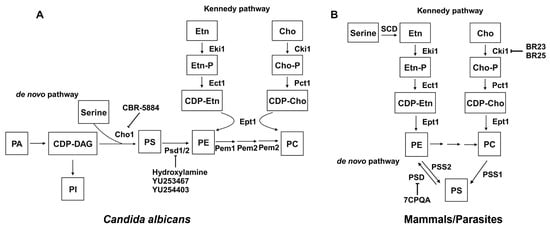
Figure 8. Phospholipid biosynthesis pathways for C. albicans (A) and mammals/parasites (B). Known inhibitors to certain targets are also shown. Both the de novo pathway and Kennedy pathway exist in each scenario. This figure is adapted and modified from [193] (under Creative Commons CC BY license). PA: phosphatidic acid; CDP-DAG: cytidine diphosphate diacylglycerol; Ser: serine; Cho1/PSS1/PSS2: PS synthase; PI: phosphatidylinositol; PS: phosphatidylserine; PE: phosphatidylethanolamine; PC: phosphatidylcholine; Etn: ethanolamine; Cho: choline; Etn-P: phosphoethanolamine; Cho-P: phosphocholine; CDP-Etn: cytidyldiphosphate-ethanolamine; CDP-Cho: cytidyldiphosphatecholine; PSD: PS decarboxylase; Eki1: ethanolamine kinase; Ect1: ethanolamine-phosphate cytidylyltransferase; Ept1: ethanolamine phosphotransferase; Cki1: choline kinase; Pct1: choline-phosphate cytidylyltransferase; SDC: serine decarboxylase; Cpt1: choline phosphotransferase.
3.3.1. PS Decarboxylases (PSD)
In both yeast and mammals, PE is synthesized through the de novo pathway by decarboxylating PS or through the Kennedy pathway by using exogenous ethanolamine (Figure 8). The Kennedy pathway contributes to the majority of PE in some mammalian cells [198,199,200], but in yeast, the majority of PE is generated by the decarboxylation of PS [194,196,201]. Also, research has shown that while the elimination of the Kennedy pathway does not impact yeast cell survival, disruption of the PSD1 gene leads to ethanolamine auxotrophy and mitochondrial instability [196]. PSDs are evolutionarily conserved across a wide range of organisms, and most are membrane-associated enzymes relying on a covalently attached pyruvoyl moiety for their activities [202]. Membrane-bound PSDs are synthesized as a single polypeptide proenzyme, which undergoes self-cleavage at a highly conserved LGST motif [203,204]. The α and β chains from the cleavage assemble into a mature PSD homodimer, with each protomer having one α and one β chain. The α chains function as the catalytic domains and β chains facilitate membrane association [202]. Recently, the structures of apo and PE-bound E. coli PSDs were solved, and structural insight into detailed mechanisms of membrane-binding, PS recognition, self-cleavage, and catalysis were provided [205,206].
Several PSD inhibitors have been proposed and are shown in Table 3.
Table 3. Potential phospholipid biosynthesis enzymes as drug targets and inhibitors.
| Compound | Target Enzyme | Organism | Reference |
|---|---|---|---|
| Hydroxylamine | PS decarboxylase | E. coli | [207,208] |
| Hydroxylamine | PS decarboxylase | S. cerevisiae and C. albicans | [209] |
| Serine hydroxamate | PS decarboxylase | E. coli | [210] |
| 7CPQA | PS decarboxylase | P. falciparum | [212] |
| YU253467 and YU254403 | PS decarboxylase | C. albicans | [120] |
| CBR-5884 | PS synthase | C. albicans | [121] |
| Validamycin A | PI synthesis | R. cerealis | [213] |
| Ethionine | PE methylation | S. cerevisiae | [214] |
| 2-hydroxyethyl-hydrazine | PE methylation | S. cerevisiae | [215] |
| BR23 and BR25 | Choline kinase | P. falciparum | [216] |
3.3.2. PS Synthase
Fungi use the Cho1 PS synthase to catalyze the formation of PS from CDP-DAG and serine, and both the enzyme and the reaction are absent in mammals [181,197,217], indicating a potential antifungal target. Furthermore, deleting the PS synthase in C. albicans prevents it from causing disease in mouse models of oral or systemic candidiasis [183,218]. PS synthase is also crucial for the growth of the major fungal pathogen C. neoformans [189] and is also highly conserved across various fungal species [217]; these observations indicate that PS synthase is an excellent drug target.
PS synthases were first characterized in bacteria. The PS synthases from Gram-negative bacteria such as E. coli, Salmonella typhimurium, and Enterobacter aerogenes are tightly associated with ribosomes, and perform catalysis when they bind to the plasma membrane [219]. Gram-positive bacteria such as B. megaterium, Bacillus subtilis, and Clostridium perfringens have membrane-associated PS synthase, which have conserved motifs and belong to the protein family that includes eukaryotic counterparts [220]. The first eukaryotic PS synthase was identified in S. cerevisiae [221,222]. Since then, the characterization of S. cerevisiae PS synthase (Cho1) included understanding the regulation of Cho1 [223,224,225,226,227], identifying the localization of the enzyme [228,229], protein solubilization and purification [230,231], and determination of Michaelis–Menton kinetics [223,230,231]. The C. albicans PS synthase was first characterized in 2010, with the finding that it is crucial for systemic Candida infections in mice [183]. Michaelis–Menton kinetics of C. albicans PS synthase were described, and its conserved CAPT motif for binding CDP-DAG was identified as well as some residues involved in serine binding [232,233,234]. Later, C. albicans PS synthase was solubilized and purified, but surprisingly formed a hexamer. It is unique among the known structures of the same family of membrane-bound phospholipid synthases, which are all dimers [235].
PS synthase belongs to the CDP-alcohol phosphatidyltransferase (CDP-AP) protein family, and six prokaryotic [236,237,238,239,240,241] and two eukaryotic [242,243] members have solved structures to date. Among these, there is only one PS synthase structure, and it is from the archaean Methanocaldococcus jannaschii, and has eight transmembrane domains [241]. This is different from the homology model of C. albicans PS synthase with six transmembrane domains. In addition, some key residues involved in catalytic activity of C. albicans PS were lacking functions in M. jannaschii PS synthase [235,241]. A structure of fungal PS synthase will reconcile this discrepancy and also provide insights into the mechanisms of C. albicans PS synthase catalysis.
Currently, despite the fact that PS synthase is a promising drug target, the identification of its specific inhibitors is in the early stage. Two cell-based screens were conducted to identify inhibitors of C. albicans PS synthase, but both attempts were unsuccessful [244,245]. One screen pinpointed the compound SB-224289. However, it was later determined that SB-224289 only acts on PS synthase-related physiological pathways rather than the enzyme directly [244]. The other screen identified bleomycin, but it was also found that this compound affects phospholipid-associated processes rather than targeting C. albicans PS synthase directly [245]. Recently, Zhou et al. identified a molecule, CBR-5884 (Table 3), that inhibits both purified PS synthase and its function in vivo, with a Ki of 1550 ± 245.6 nM [121]. This molecule acts as a competitive inhibitor for serine, thus having the potential for further development. However, more efforts are needed to identify additional inhibitors to this promising drug target that can potentially lead to new classes of antifungals.
3.3.3. Other Miscellaneous Phospholipid Synthesis Inhibitors
Finally, besides PS decarboxylase and PS synthase, several inhibitors have been identified to target phospholipid biosynthesis pathways for PI and PC. For example, it was suggested that validamycin A might hinder the incorporation of inositol into PI in Rhizoctonia cerealis, a process driven by the membrane-associated enzyme PI synthase [213], but a detailed enzymatic characterization is missing. For de novo PC biosynthesis, where PE is methylated three time into PC (Figure 8), ethionine and 2-hydroxyethyl-hydrazine were shown to inhibit PE methylation, and thus resulted in lower PC levels [214,215]. Moreover, the anticancer compounds BR23 and BR25, known to inhibit human choline kinase, directly inhibited the ethanolamine activity of P. falciparum choline kinase, thus significantly reducing PE levels in P. falciparum without affecting PC [216]. This led to halted growth of the parasite due to the depletion of membrane PE, and was ultimately lethal [216]. These observations underscore the significance of phospholipid biosynthesis in certain microbes’ survival and pathogenicity and thus drug development.
This entry is adapted from the peer-reviewed paper 10.3390/jof10030171
This entry is offline, you can click here to edit this entry!