Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, General & Internal
Peptides continue to gain significance in the pharmaceutical arena. Since the unveiling of insulin in 1921, the Food and Drug Administration (FDA) has authorised around 100 peptides for various applications. Peptides, although initially derived from endogenous sources, have evolved beyond their natural origins, exhibiting favourable therapeutic effectiveness. The FDA has authorized a variety of medications for the treatment of diabetes, each designed to target different receptors and operate through diverse mechanisms, depending on the specific type of diabetes being addressed.
- peptides
- FDA
- natural peptides
- diabetes
- insulin
1. Diabetes Type I
1.1. Insulin
2.1. Insulin (Iletin)
Insulin stands as the inaugural isolated natural peptide, serving as a peptide hormone produced and released by the β-cells located in the pancreatic islets [17]. Insulin was isolated by Frederick Banting and Best in 1921 [17,18,19]. Insulin is a 51-mer acid peptide consisting of two chains, A and B, linked by three disulfide bridges. These bridges connect Cys6 and Cys11 of chain A, Cys7 of chain A and Cys7 of chain B, and Cys20 of chain A and Cys19 of chain B (Figure 2). In 1922, insulin became the first therapeutic peptide approved for treating patients with diabetes mellitus. Eli Lilly and Company commercialised it in 1923 [19,20]. It is used to enhance glycemic control in both adults and children diagnosed with diabetes mellitus [21].
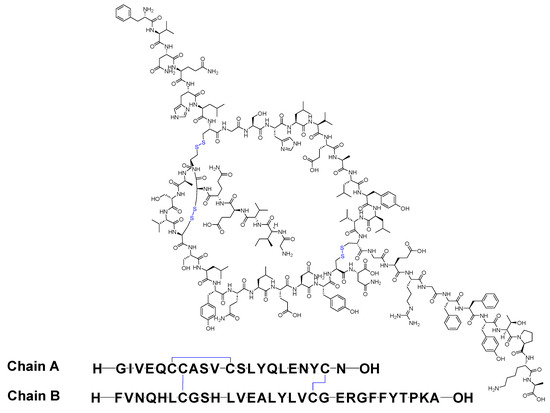
Figure 2. Chemical structure of insulin. Blue: disulfide bridges.
Insulin binds to the α subunit of the insulin receptor (INSR) located on muscle and fat cells. Upon insulin binding to the extracellular domain of the α subunits, the receptor undergoes conformational changes that activate the intrinsic tyrosine kinase activity of the β subunits. This activation leads to autophosphorylation of the β subunits and subsequent phosphorylation of downstream signalling molecules, initiating a cascade of events that regulate various cellular processes such as glucose uptake and metabolism (Figure 3) [22]. Signalling events downstream of INSR activation can be broadly categorised into mitogenic (related to cell growth and division) and metabolic signals [23,24,25]. INSR mediates a variety of cellular responses, coordinating both growth-related and metabolic processes [22]. As a result, insulin works to lower blood glucose levels by facilitating the cellular uptake of glucose and simultaneously inhibiting the release of glucose from the liver [21,22].
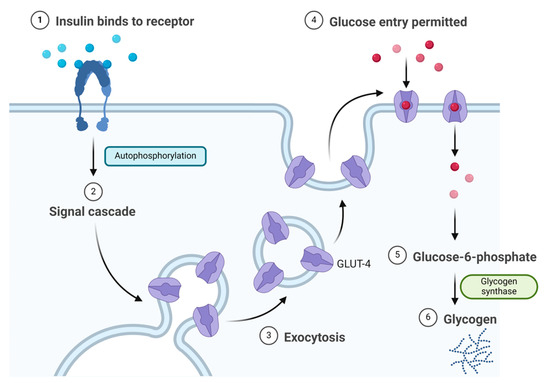
Figure 3. Insulin mechanism of action.
Eli Lilly and Company developed insulin, and it received FDA approval in 1923 [21]. Eli Lilly and Company also developed insulin lispro (Humalog), which distinguishes itself from human insulin by the substitution of lysine for proline at position B28 and proline for lysine at position B29 in its amino acid sequence [26]. It received FDA approval in 1996 [26]. Additionally, they developed insulin aspart (NovoLog), structurally similar to regular human insulin, differing only by a single substitution in which the amino acid proline is replaced by aspartic acid at position B28. This replacement in NovoLog diminishes the molecule’s inclination to form hexamers, a characteristic observed with regular human insulin. Consequently, NovoLog is absorbed more rapidly after subcutaneous injection when compared to regular human insulin [27]. It received FDA approval in 2000 [27]. It is typically administered subcutaneously and may be associated with adverse effects such as allergic reactions, local injection site reactions, lipodystrophy, rash, and pruritus [21].
1.2. Pramlintide (Symlin)
Pramlintide is a 37-mer peptide amide (KCNTATCATQRLANFLVHSSNNFGPILPPTNVGSNTY-NH2) that features a disulfide bridge between Cys2 and Cys7. It differs from the human amylin sequence by replacing Ala25, Ser28, and Ser29 with Pro in the corresponding three positions (Figure 18). It is used to treat type I and II diabetic patients treated with insulin [81].
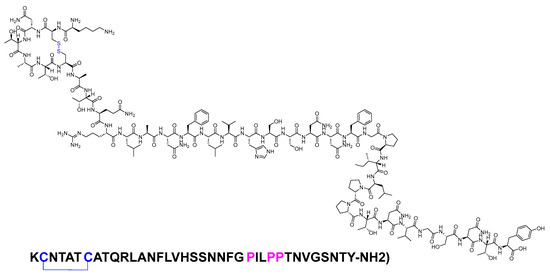
Figure 18. Chemical structure of pramlintide. Blue: disulfide bridge. Pink: positions that are different from those of natural amylin.
Pramlintide functions as an amylinomimetic agent, mimicking the actions of the hormone amylin. This includes the modulation of gastric emptying, the inhibition of postprandial glucagon secretion, and the induction of satiety, ultimately resulting in decreased caloric intake and the potential for weight loss [81,82]. Pramlintide attaches to the calcitonin receptor’s core, along with one of the three receptor activity-modifying proteins, namely RAMP1, RAMP2, or RAMP3 [81,82]. The absolute bioavailability of a single subcutaneous dose is estimated to be approximately 30 to 40%. The injection administered in the arm resulted in higher exposure compared to injections in the abdominal area or thigh. [81].
Amylin Pharmaceuticals developed pramlintide, and it was granted FDA approval in 2005 [83]. Administered subcutaneously, it exhibited the following adverse effects: headache, dizziness, drowsiness, vision problems, hunger, weakness, sweating, confusion, irritability, a fast heart rate, and a jittery feeling [81,82]. In healthy subjects, the half-life of pramlintide is approximately 48 min, and it undergoes metabolism primarily in the kidneys [81].
2. Diabetes Type 2
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP).
Both glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are key incretins released from enteroendocrine cells in the gut [84]. GIP, a 42-mer peptide, originates from K cells in the upper intestine, while GLP-1, a 31-mer peptide, is produced by L cells in the lower intestine (Figure 19) [85,86].

Figure 19. Sequences of GLP-1 and GIP. Blue represents the matching amino acid residues.
GLP-1 and GIP regulate blood glucose levels associated with meals by maintaining a balance between insulin and glucagon [85,86]. Additionally, they inhibit gastric emptying, optimising nutrient absorption, and simultaneously managing weight gain [86]. GIP and GLP-1 bind to their respective receptors, initiating a cascade of events, including the activation of adenylate cyclase, the elevation of intracellular cAMP levels, and the activation of PKA. Furthermore, cAMP2 (EPAC2)/cAMP-guanine nucleotide exchange factor (GEF)II activates the exchange of protein. PKA plays a role in closing KATP channels, leading to membrane depolarization, and inhibits the delayed rectifying K+ (Kv) channel. This depolarization results in the opening of voltage-gated Ca2+ channels (VDCC), causing an increase in intracellular Ca2+ concentrations. The elevated Ca2+ concentrations eventually trigger the fusion of insulin-containing granules with the plasma membrane, facilitating insulin secretion from the β-cells. Additionally, this process promotes the transcription of the proinsulin gene, thereby increasing the insulin content within the β-cell. Notably, the activation of EPAC2 has been demonstrated to enhance the density of insulin-containing granules near the plasma membrane, further potentiating insulin secretion from the β-cell. ATP is involved in these intricate cellular processes (Figure 20) [85].
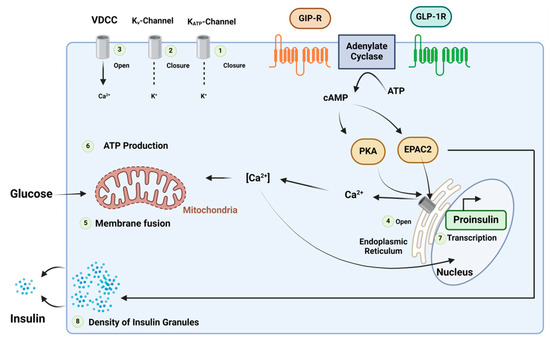
Figure 20. Mechanism of the insulinotropic effects of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide (GLP)-1. Adapted from Ref. [85].
GLP-1 and GIP analogues share a structural resemblance of approximately 90% to the native GLP-1 and GIP, making them agonists capable of initiating the insulin production process (Table 2). Notably, certain GLP-1 analogues have demonstrated relevance in cardiovascular diseases. Patients administered with GLP-1 analogues exhibited lower incidences of cardiovascular-related deaths, nonfatal myocardial infarction, or nonfatal stroke [87].
2.1. Exenatide (Byetta)
Exenatide is a 39-mer linear acid peptide (H-HGEGXFXSDLSKQMEEEAVRLFXEWLKNGGPSSGAPPPS-OH) that belongs to the GLP-1 family (Figure 21). It is used as an adjunct treatment to dietary and exercise measures to enhance glycemic control in adults diagnosed with type 2 diabetes mellitus [88].
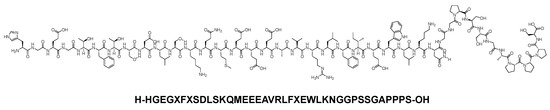
Figure 21. Chemical structure of exenatide.
Exenatide, a GLP-1 receptor agonist, binds to and activates the GLP-1 receptor, resulting in an augmentation of glucose-dependent insulin synthesis and the in vivo secretion of insulin from pancreatic β cells [88]. Similar to GLP-1 and other analogues, the mechanism involves raising the levels of cAMP, along with the activation of other downstream signalling pathways [88]. After subcutaneous administration to patients with type 2 diabetes, exenatide reaches median peak plasma concentrations in 2.1 h. Similar exposure levels are achieved with subcutaneous administration of exenatide in the abdomen, thigh, or upper arm [88].
Amylin Pharmaceuticals developed exenatide, and it received FDA approval in 2005 [89]. Administered subcutaneously, it has been associated with the following adverse effects: nausea, hypoglycaemia, vomiting, diarrhoea, jittery feelings, dizziness, headache, and dyspepsia. It is worth noting that nausea tends to decrease over time [88]. Exenatide is primarily eliminated through glomerular filtration, followed by proteolytic degradation. The mean apparent clearance of exenatide in humans is 9.1 L/h, and with a half-life of 2.4 h; it is recommended to be administered twice daily [88].
2.2. Liraglutide (Victoza)
Liraglutide is a 31-mer peptide that belongs to GLP-1 family. It possesses a hexadecanoyl-Glu-containing moiety attached to its Lys side chain (Figure 22). It is used as an adjunct treatment alongside diet and exercise to enhance glycemic control in adults diagnosed with type 2 diabetes mellitus. Additionally, it is utilised to lower the risk of major adverse cardiovascular events in adults with type 2 diabetes mellitus and established cardiovascular disease [90].
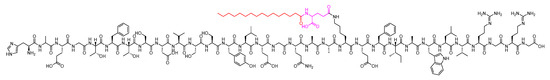
Figure 22. Chemical structure of liraglutide. Black: peptide backbone; red: hexadecanoyl; pink: Glu.
Liraglutide, functioning as a GLP-1 receptor agonist, binds to and activates the GLP-1 receptor. This activation leads to an elevation in intracellular cAMP, thereby promoting insulin secretion when confronted with elevated glucose levels. Additionally, liraglutide exhibits the capability to reduce glucagon levels in a glucose-dependent manner. The overall process also encompasses a delay in gastric emptying [90]. The self-association and delayed absorption, coupled with its binding to plasma proteins and stability against metabolic degradation by dipeptidyl peptidase IV (DPP-IV) and neutral endopeptidase (NEP), contribute to a pharmacokinetic profile that renders it well-suited for once-daily administration [90]. Following subcutaneous administration, maximum concentrations of liraglutide are achieved at 8–12 h post dosing [90].
Liraglutide was developed by Novo Nordisk and received FDA approval in 2010 [91]. Administered subcutaneously, it may lead to the following adverse effects: nausea, diarrhoea, vomiting, decreased appetite, dyspepsia, and constipation [90]. In the first 24 h after the administration of a single dose of liraglutide to healthy subjects, the primary component in plasma was intact liraglutide. Liraglutide undergoes endogenous metabolism in a manner similar to that of large proteins, with no specific organ identified as a major route of elimination [90].
2.3. Lixisenatide (Adlyxin)
Lixisenatide is a 44-mer peptide amide (H-HGEGTFTSDLSKQMEEEAVRLFIEWLKNGGPSSGAPPSKKKKKK-NH2) that belongs to the GLP-1 family (Figure 23). Lixisenatide was derived from exenatide, with a modification involving the replacement of a Pro residue from the C-terminal with a linker comprising six Lys residues [92]. With its six Lys residues, it exhibits a heightened resistance to degradation by DPP-IV, thereby extending its half-life and facilitating once-daily dosing [93].
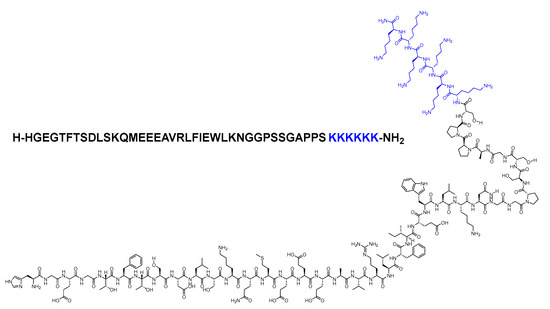
Figure 23. Chemical structure of lixisenatide. Blue: difference from exenatide.
It is used as an adjunct treatment alongside diet and exercise to enhance glycemic control in adults diagnosed with type 2 diabetes mellitus [94].
Lixisenatide, functioning as a GLP-1 receptor agonist, binds to and activates this receptor, thereby stimulating adenylyl cyclase. This activation results in increased glucose-dependent insulin secretion, decreased glucagon secretion, and a slowing of gastric emptying [86,94].
It was developed by Sanofi Aventis and approved by the FDA in 2013 [95]. Administered subcutaneously, it may elicit the following side effects: nausea, vomiting, headache, diarrhoea, dizziness, and hypoglycaemia [94]. Lixisenatide was discontinued in 2023, with the decision attributed solely to business considerations and not related to any concerns about the safety or efficacy of the drug [96].
2.4. Albiglutide (Tanzeum)
Albiglutide consists of two copies of a 30-mer linear acid peptide (H-HGEGTFTSDVSSYLEGQAAKEFIAWLVKGRG-OH), and it belongs to GLP-1 family (Figure 24). It is used as an adjunct treatment alongside diet and exercise to enhance glycemic control in adults diagnosed with type 2 diabetes mellitus [97].
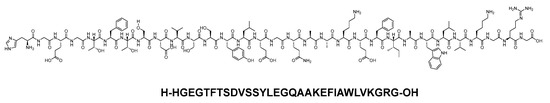
Figure 24. Chemical structure of abiglutide.
Abiglutide, functioning as a GLP-1 receptor agonist, activates the GLP-1 receptor, leading to increased glucose-dependent insulin secretion, and it also has the effect of slowing gastric emptying [98].
It was developed by GSK and received FDA approval in 2014 [99]. However, in 2017, it was discontinued primarily due to limited prescribing of the drug and not because of any safety or efficacy concerns [98]. Administered subcutaneously, it may result in the following adverse effects: upper respiratory tract infection, diarrhoea, nausea, injection site reaction, cough, back pain, arthralgia, sinusitis, and influenza [97].
2.5. Dulaglutide (Trulicity)
Dulaglutide is a 31-mer linear acid peptide (H-HGEGTFTSDVSSYLEEQAAKEFIAWLVKGGG-OH) that belongs to GLP-1 family (Figure 25). It is used alongside diet and exercise to enhance glycemic control in adults diagnosed with type 2 diabetes mellitus. Additionally, it is utilised to lower the risk of major adverse cardiovascular events in adults with type 2 diabetes mellitus who have established cardiovascular disease or multiple cardiovascular risk factors [100].
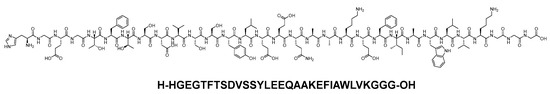
Figure 25. Chemical structure of dulaglutide.
Dulaglutide, a GLP-1 receptor agonist, binds to and activates the GLP-1 receptor, resulting in an augmentation of glucose-dependent insulin synthesis and the in vivo secretion of insulin from pancreatic β cells [100]. Dulaglutide elevates the levels of cAMP, along with engaging other downstream signalling pathways. This process culminates in the glucose-dependent release of insulin, a reduction in glucagon secretion, and a deceleration of gastric emptying [100,101]. After subcutaneous administration, the time to reach the maximum plasma concentration of dulaglutide at a steady state varies from 24 to 72 h, with a median of 48 h. Following once-weekly administration, steady-state plasma dulaglutide concentrations are achieved between 2 and 4 weeks, and the accumulation ratio is approximately 1.56. Importantly, the site of subcutaneous administration (abdomen, upper arm, and thigh) does not have a statistically significant effect on the exposure to dulaglutide [100].
Dulaglutide was developed by Eli Lilly and Company and received FDA approval in 2014 [102,103]. Administered subcutaneously, it has been associated with the following adverse effects: nausea, diarrhoea, vomiting, abdominal pain, and decreased appetite [100]. Dulaglutide exhibited an apparent population mean clearance of 0.142 L/h, with an elimination half-life of around 5 days [100]. Dulaglutide is presumed to undergo degradation into its component amino acids through general protein catabolism pathways [100].
2.6. Semaglutide (Ozempic)
Semaglutide is a 31-mer acid peptide that belongs to GLP-1 family. It possesses a Glu-containing moiety appended to its Lys side chain, along with two mini-PEG amino acids (8-amino-3,6-dioxaoctanoic acid, ADO) and a C18 diacid. The inclusion of the alpha-aminoisobutyric acid (Aib) residue contributes to the enhanced stability of the peptide against DDP-IV (Figure 26) [104,105,106]. It is as an adjunct treatment alongside diet and exercise to enhance glycemic control in adults diagnosed with type 2 diabetes mellitus [107].
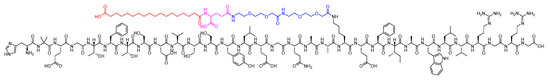
Figure 26. Chemical structure of semaglutide. Black: peptide backbone; red: 17-carboxyheptadecanoyl (C18 diacid); pink: Glu; blue: 8-amino-3,6-dioxaoctanoic acid (ADO).
Semaglutide, as a GLP-1 receptor agonist, binds to and activates the GLP-1 receptor. Similar to the actions of GLP-1, it carries out several effects on glucose regulation through the activation of the GLP-1 receptor [108]. It stimulates the insulin secretion and inhibits the glucagon secretion in a glucose-dependent manner [107]. It exhibits extensive binding to albumin protein, enabling a once-weekly administration [107]. The absolute bioavailability of semaglutide is 89%. The maximum concentration of semaglutide is typically reached 1 to 3 days post-dose. Additionally, similar exposure levels are attained with subcutaneous administration of semaglutide in the abdomen, thigh, or upper arm [107].
Semaglutide was developed by Novo Nordisk and received FDA approval in 2017 [109]. Administered subcutaneously, it may lead to side effects such as nausea, vomiting, diarrhoea, abdominal pain, and constipation [107]. The main route of elimination for semaglutide is through metabolism, involving proteolytic cleavage of the peptide backbone and sequential beta-oxidation of the fatty acid side chain [107].
2.7. Tirzepatide (Mounjaro)
Tirzepatide is a 39-mer peptide amide, functioning as a GLP-1 analogue, and it incorporates a C20 fatty acid diacidic moiety. This structural feature facilitates binding to albumin, thereby extending its half-life [110]. The inclusion of two Aib nonproteinogenic residues contributes to the stability of the peptide by protecting it against peptidase degradation (Figure 27) [92]. It is used as a treatment, in conjunction with diet and exercise, to enhance glycemic control in adults diagnosed with type 2 diabetes mellitus [110].
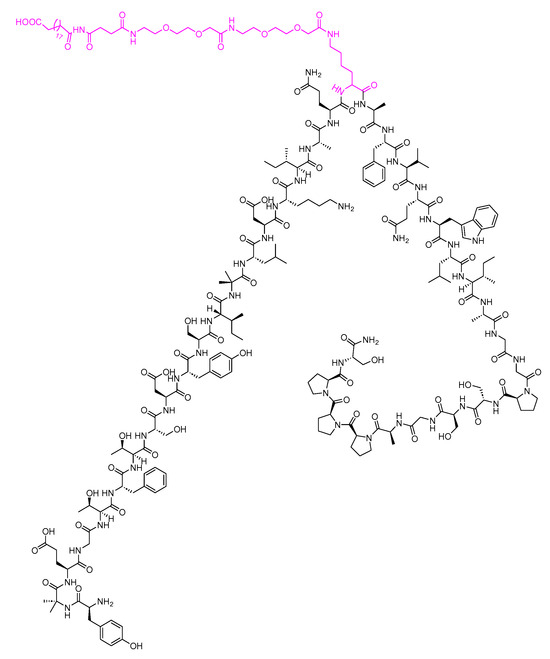
Figure 27. Chemical structure of tirzepatide. Pink: C20 fatty acid diacidic moiety.
Tirzepatide acts as an agonist for both GLP-1 and GIP [111]. Tirzepatide selectively binds to both GIP and GLP-1 receptors, activating them. This activation enhances the phases of insulin secretion and reduces glucagon levels in a glucose-dependent manner [110]. After subcutaneous administration, the time to reach maximum plasma concentration of tirzepatide varies from 8 to 72 h. The mean absolute bioavailability of tirzepatide following subcutaneous administration is 80%. Additionally, similar exposure levels are achieved with subcutaneous administration of tirzepatide in the abdomen, thigh, or upper arm. [110].
Tirzepatide was developed by Eli Lilly and Company and received FDA approval in 2022 [112]. Administered subcutaneously, tirzepatide has been associated with various adverse effects, including nausea, diarrhoea, decreased appetite, vomiting, constipation, dyspepsia, and abdominal pain [110]. The apparent population mean clearance of tirzepatide is 0.061 L/h, and it has an elimination half-life of approximately 5 days. This prolonged half-life allows for once-weekly dosing [110]. Tirzepatide undergoes metabolism through proteolytic cleavage of the peptide backbone, beta-oxidation of the C20 fatty diacid moiety, and amide hydrolysis [110].
This entry is adapted from the peer-reviewed paper 10.3390/biom14030264
This entry is offline, you can click here to edit this entry!