Unlike classic antiphospholipid syndrome (APS), catastrophic antiphospholipid syndrome (CAPS) causes multiple microthrombosis due to an increased inflammatory response, known as a “thrombotic storm”. CAPS typically develops after infection, trauma, or surgery and begins with the following symptoms: fever, thrombocytopenia, muscle weakness, visual and cognitive disturbances, abdominal pain, renal failure, and disseminated intravascular coagulation. Although the presence of antiphospholipid antibodies in the blood is one of the diagnostic criteria, the level of these antibodies can fluctuate significantly, which complicates the diagnostic process and can lead to erroneous interpretation of rapidly developing symptoms.
- antiphospholipid antibodies
- antiphospholipid syndrome
- complications of pregnancy
- catastrophic antiphospholipid syndrome
1. Introduction
2. Risk Factors and Pathogenesis
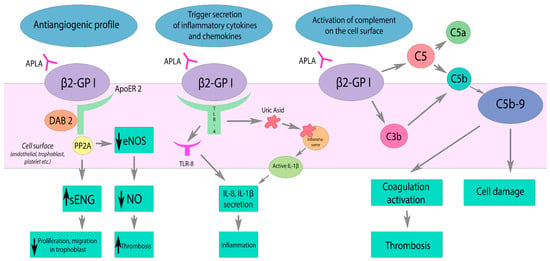
- (1)
-
inhibition of PC complex assembly,
- (2)
-
reducing the activation of PC through the thrombomodulin–thrombin complex,
- (3)
-
direct suppression of PC activity,
- (4)
-
decreased clearance of PC.
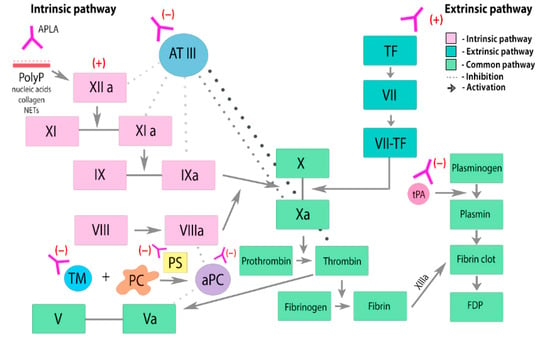
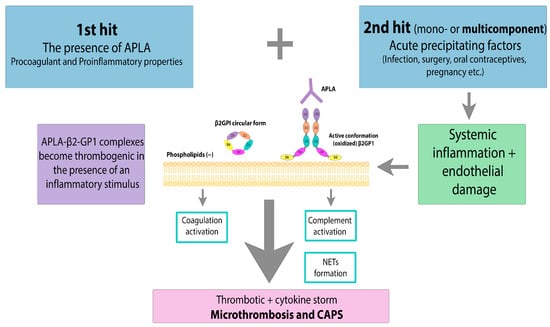
3. Clinical Manifestations
4. Diagnostic Difficulties
- (1)
-
Body temperature more than 38 °C or less than 36 °C;
- (2)
-
Pulse rate more than 90/min;
- (3)
-
Respiratory rate more than 20/min or PaCO2 less than 32 mmHg (4.3 kPa);
- (4)
-
Laboratory tests: white blood cell count more than 12,000/mL or less than 4000/mL or more than 10% immature cells [35].
5. Principles of Treatment
- (1)
-
Trigger elimination (termination of pregnancy, treatment of infection, etc.) if possible;
- (2)
-
Full anticoagulation with LMWH at a dose of 1 mg/kg (in the case of enoxaparin) every twelve hours;
- (3)
-
Glucocorticoids;
- (4)
-
Plasmapheresis with plasma exchange.
6. Prognosis and Prevention
6.1. Prognosis
6.2. Prevention
6.3. CAPS and SARS-CoV-2
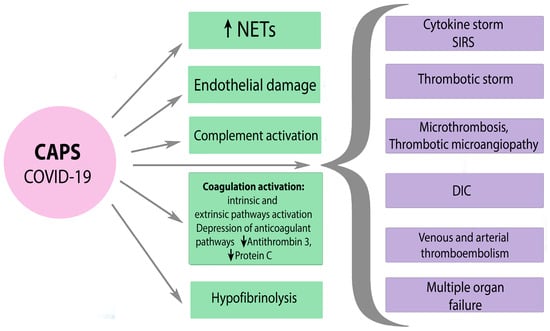
7. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/ijms25010668
References
- Vora, S.K.; Asherson, R.A.; Erkan, D. Catastrophic antiphospholipid syndrome. J. Intensive Care Med. 2006, 21, 144–159.
- Bitsadze, V.O.; Khizroeva, D.K.; Makatsariya, N.A.; Egorova, E.S.; Baymuradova, S.M.; Mashkova, T.Y. Antiphospholipid anti-bodies, their pathogenetic and diagnostic issues obstetric practice. Obstet. Gynecol. Reprod. 2014, 8, 39–60.
- Asherson, R.A.; Cervera, R.; De Groot, P.G.; Erkan, D.; Boffa, M.-C.; Piette, J.-C.; Khamashta, M.A.; Shoenfeld, Y.; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: International consensus statement on classification criteria and treatment guidelines. Lupus 2003, 12, 530–534.
- Asherson, R.A. The catastrophic antiphospholipid syndrome. J. Rheumatol. 1992, 19, 508–512.
- Cervera, R. CAPS Registry. Lupus 2012, 21, 755–757.
- Cervera, R.; Espinosa, G. Update on the catastrophic antiphospholipid syndrome and the ‘CAPS Registry’. Semin. Thromb. Hemost. 2012, 38, 333–338.
- Cervera, R.; Rodríguez-Pintó, I.; Legault, K.; Erkan, D. 16th International Congress on Antiphospholipid Antibodies Task Force Report on Catastrophic Antiphospholipid Syndrome. Lupus 2020, 29, 1594–1600.
- Makatsariya, A.; Asherson, R.; Bitsadze, V.; Baimuradova, S.; Akinshina, S. Catastrophic antiphospholipid (Asherson’s) syndrome and genetic thrombophilic disorders in obstetrics. Autoimmun. Rev. 2006, 6, 89–93.
- Tektonidou, M.G.; Andreoli, L.; Limper, M.; Amoura, Z.; Cervera, R.; Costedoat-Chalumeau, N.; Cuadrado, M.J.; Dörner, T.; Ferrer-Oliveras, R.; Hambly, K.; et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann. Rheum. Dis. 2019, 78, 1296–1304.
- Pregnolato, F.; Gerosa, M.; Raimondo, M.G.; Comerio, C.; Bartoli, F.; Lonati, P.A.; Borghi, M.O.; Acaia, B.; Ossola, M.W.; Ferrazzi, E.; et al. EUREKA algorithm predicts obstetric risk and response to treatment in women with different subsets of anti-phospholipid antibodies. Rheumatology 2021, 60, 1114–1124.
- Giannakopoulos, B.; Krilis, S.A. The pathogenesis of the antiphospholipid syndrome. N. Engl. J. Med. 2013, 368, 1033–1044.
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339.
- Ağar, Ç.; van Os, G.M.; Mörgelin, M.; Sprenger, R.R.; Marquart, J.A.; Urbanus, R.T.; Derksen, R.H.; Meijers, J.C.; de Groot, P.G. Beta2-Glycoprotein I can exist in 2 conformations: Implications for our understanding of the antiphospholipid syndrome. Blood 2010, 116, 1336–1343.
- DE Groot, P.G.; Meijers, J.C.M. β(2)-Glycoprotein I: Evolution, structure and function. J. Thromb. Haemost. 2011, 9, 1275–1284.
- Agostinis, C.; Biffi, S.; Garrovo, C.; Durigutto, P.; Lorenzon, A.; Bek, A.; Bulla, R.; Grossi, C.; Borghi, M.O.; Meroni, P.; et al. In vivo distribution of β2 glycoprotein I under various pathophysiologic conditions. Blood 2011, 118, 4231–4238.
- Di Simone, N.; Di Nicuolo, F.; D’Ippolito, S.; Castellani, R.; Tersigni, C.; Caruso, A.; Meroni, P.; Marana, R. Antiphospholipid antibodies affect human endometrial angiogenesis. Biol. Reprod. 2010, 83, 212–219.
- Martínez-Flores, J.A.; Serrano, M.; Serrano, A. Antiphospholipid immune complexes as thrombosis risk marker. Oncotarget 2019, 10, 805–806.
- Sacharidou, A.; Chambliss, K.L.; Ulrich, V.; Salmon, J.E.; Shen, Y.M.; Herz, J.; Hui, D.Y.; Terada, L.S.; Shaul, P.W.; Mineo, C. Thrombosis and Hemostasis: Antiphospholipid antibodies induce thrombosis by PP2A activation via apoER2-Dab2-SHC1 complex formation in endothelium. Blood 2018, 131, 2097.
- Romay-Penabad, Z.; Aguilar-Valenzuela, R.; Urbanus, R.T.; Derksen, R.H.; Pennings, M.T.; Papalardo, E.; Shilagard, T.; Vargas, G.; Hwang, Y.; De Groot, P.G.; et al. Apolipoprotein E receptor 2 is involved in the thrombotic com-plications in a murine model of the antiphospholipid syndrome. Blood 2011, 117, 1408.
- Urbanus, R.; de Laat, B. Antiphospholipid antibodies and the protein C pathway. Lupus 2010, 19, 394–399.
- de Laat, B.; Derksen, R.H.W.M.; Mackie, I.J.; Roest, M.; Schoormans, S.; Woodhams, B.J.; de Groot, P.G.; van Heerde, W.L. Annexin A5 polymorphism (-1C->T) and the presence of anti-annexin A5 antibodies in the antiphospholipid syndrome. Ann. Rheum. Dis. 2006, 65, 1468–1472.
- Rand, J.; Wu, X.-X.; Quinn, A.; Taatjes, D. The annexin A5-mediated pathogenic mechanism in the antiphospholipid syndrome: Role in pregnancy losses and thrombosis. Lupus 2010, 19, 460–469.
- Forastiero, R.; Martinuzzo, M. Prothrombotic mechanisms based on the impairment of fibrinolysis in the antiphospholipid syndrome. Lupus 2008, 17, 872–877, Erratum in Lupus 2009, 18, 93.
- Ruffatti, A.; Tonello, M.; Macor, P.; Calligaro, A.; Del Ross, T.; Favaro, M.; Lotti, V.; Carletto, A.; Hoxha, A.; Biasi, D. Markers of complement activation in plasma during quiescent phases in patients with catastrophic antiphospholipid syndrome. Blood 2021, 137, 2989–2992.
- Ruffatti, A.; Tarzia, V.; Fedrigo, M.; Calligaro, A.; Favaro, M.; Macor, P.; Tison, T.; Cucchini, U.; Cosmi, E.; Tedesco, F.; et al. Evidence of complement activation in the thrombotic small vessels of a patient with catastrophic antiphospholipid syndrome treated with eculizumab. Autoimmun. Rev. 2019, 18, 561–563.
- Rodríguez-Pintó, I.; Moitinho, M.; Santacreu, I.; Shoenfeld, Y.; Erkan, D.; Espinosa, G.; Cervera, R. Catastrophic antiphospholipid syndrome (CAPS): Descriptive analysis of 500 patients from the International CAPS Registry. Autoimmun. Rev. 2016, 15, 1120–1124.
- Sanchez, J.M.; Davila, M.; Amer, R. Catastrophic Antiphospholipid Syndrome: Skin, Eye and Brain Involvement. Ocul. Immunol. Inflamm. 2022, 30, 986–988.
- Oredegbe, A.A.; Robledo, F.M.S.; Hongalgi, K.; Faddoul, G.; Mehta, S. Bilateral adrenal hemorrhage: A rare presentation of catastrophic an-ti-phospholipid syndrome. Am. J. Med. Sci. 2023, 365, 104–108.
- de Carvalho, J.F.; Silva, F.F.; Shoenfeld, Y. Pediatric catastrophic antiphospholipid syndrome patient evolving to systemic lupus erythematosus. Lupus 2021, 30, 155–157.
- Huang, C.; Zhao, Y.; Tian, X.; Wang, Q.; Hu, C.; Jiang, N.; Zhou, S.; Zhang, L.; Zhou, J.; Wu, C.; et al. Early recognition of catastrophic antiphospholipid syndrome in patients with antiphospholipid syndrome based on a Chinese cohort study. Clin. Exp. Rheumatol. 2023, 41, 1017–1023.
- Rosário, C.; Zandman-Goddard, G.; Meyron-Holtz, E.G.; D’cruz, D.P.; Shoenfeld, Y. The Hyperferritinemic Syndrome: Macrophage activation syndrome, Still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. 2013, 11, 185.
- Egiziano, G.; Widdifield, J.; Rahman, A.; Vinet, E.; Moura, C.S.; Curtis, J.R.; Bernatsky, S. Antiphospholipid Antibody Testing in a General Population Sample from the USA: An Administrative Database Study. Sci. Rep. 2020, 10, 3102.
- Avčin, T.; Toplak, N. Antiphospholipid antibodies in response to infection. Curr. Rheumatol. Rep. 2007, 9, 212–218.
- Wenzel, C.; Stoiser, B.; Locker, G.J.; Laczika, K.; Quehenberger, P.; Kapiotis, S.; Frass, M.; Pabinger, I.; Knöbl, P. Frequent development of lupus anticoagulants in critically ill patients treated under intensive care conditions. Crit. Care Med. 2002, 30, 763–770.
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810.
- Espinosa, G.; Cervera, R.; Asherson, R.A. Catastrophic antiphospholipid syndrome and sepsis. A common link? J. Rheumatol. 2007, 34, 923–926.
- Kim, S.; Moskowitz, N.K.; DiCarlo, E.F.; Bass, A.R.; Erkan, D.; Lockshin, M.D. Catastrophic Antiphospholipid Syndrome Triggered by Sepsis. HSS J. 2009, 5, 67–72.
- Tucker, S.A.; Choi, J.; Khullar, D. A probable case of catastrophic antiphospholipid syndrome: Should high-dose steroids be given in the setting of polymicrobial sepsis? SAGE Open Med. Case Rep. 2019, 7, 2050313X19839531.
- Kravitz, M.S.; Shoenfeld, Y. Thrombocytopenic conditions-autoimmunity and hypercoagulability: Commonalities and differences in ITP, TTP, HIT, and APS. Am. J. Hematol. 2005, 80, 232–242.
- Hoppensteadt, D.A.; Walenga, J.M. The relationship between the antiphospholipid syndrome and heparin-induced thrombocytopenia. Hematol. Clin. N. Am. 2008, 22, 1–18.
- Barcellona, D.; Melis, M.; Floris, G.; Mameli, A.; Muroni, A.; Defazio, G.; Marongiu, F. A “Catastrophic” Heparin-Induced Thrombocytopenia. Case Rep. Med. 2020, 2020, 6985020.
- George, D.; Vasanth, L.; Erkan, D.; Bass, A.; Salmon, J.; Lockshin, M.D. Primary Antiphospholipid Syndrome Presenting as HELLP Syndrome. HSS J. 2007, 3, 216–221.
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589.
- Legault, K.; Schunemann, H.; Hillis, C.; Yeung, C.; Akl, E.A.; Carrier, M.; Cervera, R.; Crowther, M.; Dentali, F.; Erkan, D.; et al. McMaster RARE-Bestpractices clinical practice guideline on diagnosis and management of the catastrophic antiphospholipid syndrome. J. Thromb. Haemost. 2018, 16, 1656–1664.
- Bonaventura, A.; Vecchié, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329.
- Cárdenas Suri, H.; Jimomila Bening, D. Catastrophic antiphospholipid antibody syndrome and multiple organ dysfunctions in critically ill patients with COVID-19. Expert. Rev. Respir. Med. 2020, 14, 1071–1072.