Unlike classic APS, CAPS causes multiple microthrombosis due to an increased inflammatory response, known as a “thrombotic storm”. CAPS typically develops after infection, trauma, or surgery and begins with the following symptoms: fever, thrombocytopenia, muscle weakness, visual and cognitive disturbances, abdominal pain, renal failure, and disseminated intravascular coagulation. Although the presence of antiphospholipid antibodies in the blood is one of the diagnostic criteria, the level of these antibodies can fluctuate significantly, which complicates the diagnostic process and can lead to erroneous interpretation of rapidly developing symptoms.
1. Introduction
APS remains one of the most mysterious syndromes in modern medicine. APS is a heterogeneous systemic syndrome and at the same time an autoimmune disease, acquired immune thrombophilia, and thromboinflammatory disease.
Clinical manifestations are caused by antibody-mediated activation of key target cells and modulation of several major biological systems through the interaction of these antibodies with various cofactors and special cell surface receptors. Such interactions lead to the activation of complement on cell surfaces, activation of neutrophils and monocytes, release of antiangiogenic factors, reactive oxygen species (ROS), TNF-a, activation of blood coagulation, inflammation, and NETosis (neutrophil extracellular trap formation). Because of the variable clinical manifestations of APS, it is often called “chameleon syndrome”. However, not all clinical manifestations are included in the criteria for APS, nor are antiphospholipid antibodies, which are divided into criteria (lupus anticoagulant (LA), anticardiolipin (ACL), anti-β2-glycoprotein I (anti-β2GPI)) and non-criteria (antiannexin A2, antivimentin/cardiolipin complex, antiannexin A5, antiphosphatidylethanolamine, antiphosphatidylinositol, etc.). Therefore, the classification criteria for APS are revised from time to time.
Catastrophic antiphospholipid syndrome (CAPS) is a rare and life-threatening condition, characterized by thromboses with the development of multiple organ failure [
2]. The classification criteria for CAPS are: damage to three or more organs, rapid development of clinical manifestations, histopathological patterns of small vessel occlusion and the circulating APLA [
3,
4]. The disease was first described by S. Greisman in 1991. However, this pathology is also called Asherson syndrome in honor of the doctor who first introduced the term “CAPS” in 1992 [
5]. In the 1980s, while working at the Hammersmith hospital, Ronald Asherson began collecting and describing rare cases of CAPS. Together with Ricardo Cervera, they initiated the creation of a worldwide database. The result of their work was the CAPS international register, where everyone can add clinical cases [
6]. Although CAPS develops in less than 1% of patients with APS, it is a life-threatening condition that requires early diagnosis and immediate treatment [
7].
2. Risk Factors and Pathogenesis
Extreme care and vigilance are required to ensure that the disease is not missed if a patient has risk factors for CAPS. The main risk factors include infection, surgery or trauma, anticoagulant withdrawal, pregnancy and postpartum period, combined oral contraceptive intake, vaccination, ovulation induction in ART, and many other factors [
8]. Genetic thrombophilia in pregnant women with APS is also a significant risk factor for the development of CAPS, especially in the 3rd trimester and in the postpartum period [
9]. The European League Against Rheumatism (EULAR) recommendations defined a high-risk thrombotic profile as the presence of LA or double/triple APLA positivity or the presence of persistently high APLA titers [
10]. However according to the re-evaluated risk profile of the obstetric APS, low APLA titers confer risk for pregnancy morbidity in addition to the widely accepted risk associated with medium/high APLA titers; persistent double positivity for LA and low titers of anti-β2GPI IgG display a higher risk than low aCL and anti-β2GPI IgG, either alone or associated [
11].
APLA affects many elements of the hemostatic system, and the main pathogenetic mechanisms can be divided into four groups:
- (1)
-
cellular activation (endothelial, immune cells, platelets),
- (2)
-
inhibition of anticoagulant potential,
- (3)
-
inhibition of fibrinolysis,
- (4)
-
activation of complement system [
12,
13] (
Figure 1).
Figure 1. Effects of APLA on the complement system, inflammation, and vascular tone. β2GP1—β2-glycoprotein I, ApoER2—apolipoprotein E2 receptor, DAB2—Disabled-2, PP2A—Protein phosphatase 2A, eNOS—nitric oxide synthase, sENG—soluble Endoglin, TLR-4—toll-like receptor 4, TLT-8—toll-like receptor 8. APLA reduces eNOS activity via ApoER2 interaction. Decreased NO production results in impaired vasodilatation and endothelial dysfunction. APLA activates TLR and inflammasome pathways, triggering the secretion of inflammatory cytokines and chemokines. APLA activates complement on the cell surface, leading to the coagulation activation and cell damage by C5b-9 deposition. Black arrow down—decrease; black arrow up—increase.
As a result of the presence of APLA, stimulation of endothelial and immune cells, as well as platelets, occurs. The interaction of APLA with anionic determinants (phospholipids of cell surfaces of endothelial cells, throphoblasts, etc.; polyphosphates of platelets or nucleic acids, etc.) requires the presence of β2GPI. β2GPI is the principal cofactor of APLA and exists in two different conformations: circular and open (activated) [
14]. In the circular conformation, the epitope is protected from the plasma, but after interaction with anionic surfaces the epitope becomes exposed. The formation of antibodies with these epitopes is crucial in the pathogenesis of APS and thrombogenesis [
15]. The distribution of β2GPI has distinct features. It is not present on resting endothelium but can be found on the vascular endothelium only after an inflammatory stimulus. However, it is present on the endothelium of uterine vessels and trophoblast, as well as in placental tissues at the implantation site in a normal pregnancy [
16,
17,
18].
As a result of p38 mitogen-activated kinase (p38-MAPK) and the activation of the transcription factor NF-κB, a proinflammatory and prothrombotic response develops. It manifests by an expression of adhesion molecules (E-selectin, ICAM-1, VCAM-1) and the production of cytokines (TNF-a, IL-1b, IL-6). Cytokines activate lymphocyte adhesion that contributes to further progression of inflammation. APLA also leads to decreased activity of endothelial nitric oxide synthase (eNOS). Interacting with the apolipoprotein E2 receptor (ApoER2), APLA results in decreased nitric oxide production. NO deficiency leads to impaired vasodilation and promotes platelet adhesion [
19]. Circulating anti-β2GPI also leads to activation and increased expression of tissue factor (TF) on the surface of endothelial cells and monocytes. TF, composed of the apolipoprotein C-III and a phospholipid complex, is a cell surface receptor and cofactor for coagulation factor VII (FV VII) [
20].
APLA’s persistence also leads to the inhibition of endogenous anticoagulants such as protein C (PC), protein S, prothrombin, and annexin [
21,
22]. Activation of PC occurs due to the binding of thrombomodulin and thrombin on the surface of endothelial cells and platelets. APLA inhibits protein C by:
- (1)
-
inhibition of PC complex assembly,
- (2)
-
reducing the activation of PC through the thrombomodulin–thrombin complex,
- (3)
-
direct suppression of PC activity,
- (4)
-
decreased clearance of PC.
Annexin V, which has a high affinity for phospholipids (PLs), is a natural anticoagulant and a marker of apoptosis. The phospholipid membranes of apoptotic cells are bound to annexin V, that prevents the formation of autoantibodies and a possible excessive procoagulant response. APLA can destroy the “annexin shield” when competing with PLs such as phosphatidylserine, that provokes the development of an autoimmune response [
23].
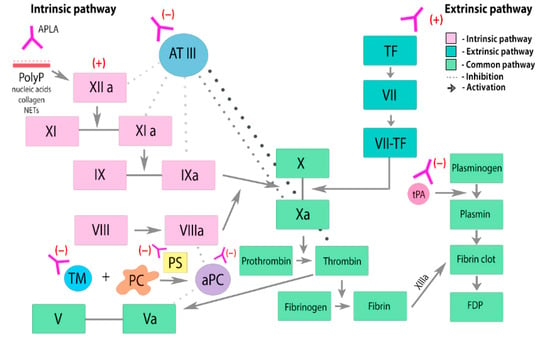
APLA can also influence the extrinsic and intrinsic fibrinolysis pathways (
Figure 2). Excessive activity of the coagulation system is regulated by complex interactions of activators and inhibitors, cofactors, and receptors. Plasminogen is converted to plasmin through the action of tissue (tPA) and urokinase (uPA) plasminogen activators. Plasminogen activator inhibitor-1 (PAI-1) inhibits the activity of both tPA and uPA. APLA leads to endothelial activation, resulting in the release of increased amounts of tPA and PAI-1. By interacting with annexin A2 (AnnA2), APLA prevents tPA from binding to its endothelial receptor. APLA potentially interferes with the normal function of each of the fibrinolytic proteins, contributing to the hypofunction of the fibrinolytic system [
24,
25].
Figure 2. Effects of APLA on the coagulation cascade and fibrinolysis. PC—protein C. PS—protein S. aPC—activated protein C. TF—tissue factor. ATIII—antithrombin III. PolyP—anionic polyphosphates. TM—thrombomodulin. APLA prevents thrombin inactivation by inhibiting antithrombin III. APLA may interfere with the components of protein C activation pathway and inhibit its anticoagulant effect, leading to an increased risk of thrombosis. APLA inhibits tPA-mediated conversion of plasminogen to plasmin, leading to dysregulation and suppression of fibrinolysis.
The complement system is also involved in the pathogenesis mechanism. In patients with the acute phase of the disease, the levels of complement C3 and C4 are decreased, but complement activation products, for example, C5b-9, are consequently increased [
26]. Complement activation mainly occurs by the classical pathway. As a result of the action of C3 convertase, the C3 component is split into C3a and C3b. On the surface of platelets, C3a binds to its receptor. As a result, platelet activation, adhesion, and aggregation occur. C3b promotes phagocytosis and is involved in the assembly of C5 convertase, which cleaves C5 into C5a and C5b. C5a stimulates the expression of tissue factor (monocytes, neutrophils, endothelial cells) and PAI-1 (mast cells, basophils). C5b is involved in the assembly of the membrane attack complex (C5b-9) and its deposition on the cell surface, which leads to the cell damage.
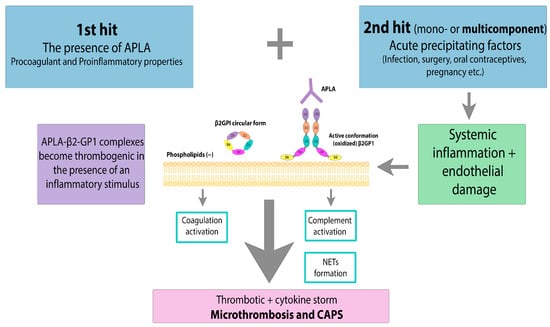
However, there is still debate about why CAPS occurs even in the presence of long-term APLA. The “two-hit” theory suggests that there must be an additional biological triggering or precipitating factors leading to endothelial activation, massive cytokine release, microvasculopathy, and thrombosis (Figure 3).
Figure 3. CAPS: the “two-hit” theory. APLA—antiphospholipid antibody, β2GPI—β2-glycoprotein I. Acute precipitating factors as a 2nd hit lead to systemic inflammation and endothelial damage. Massive cytokine release, NET formation and coagulation activation result in microthrombosis and CAPS.
3. Clinical Manifestations
According to a descriptive analysis of 500 patients included in the registry, CAPS was more common in women (69%). Forty percent of patients had associated autoimmune diseases such as SLE. The majority of CAPS episodes were triggered by various factors, with infection being the most frequent one (49%). Almost all organs and systems can be involved in CAPS manifestation. However, the skin, lungs, kidneys, central nervous system, heart, and gastrointestinal organs are the most affected [
35].
Sanchez et al. [
37] described a case of CAPS involving the skin, eyes, and brain. A 17-year-old patient developed fatigue, loss of appetite, ulcerative skin lesions of the lower extremities, and livedo reticularis. The results of an ophthalmological examination revealed ischemic damage to the retina in the temporal quadrant, as well as perivascular hemorrhages of the ocular fundus. MRI of the brain showed several lacunar white matter infarcts and small cortical/subcortical and subarachnoid hemorrhages. After 2 months of intensive therapy, the patient’s condition improved.
Oredegbe et al. [
39] presented a difficult-to-diagnose case of CAPS in a 51-year-old woman with a fulminant type of disease. The patient was initially hospitalized with abdominal pain, and a full examination revealed hemorrhage in the adrenal glands and multiple thromboses affecting many organ systems. This clinical case is considered particularly difficult to diagnose, since bilateral adrenal hemorrhage was the first manifestation of CAPS in the patient. To ensure successful treatment of this condition, it is crucial to consider the possibility of CAPS in patients presenting with multiple organ failure, multiple thromboses, and bleeding.
However, CAPS does not always develop in patients with diagnosed SLE. Carvalho et al. [
44] described the case of a 13-year-old girl who was admitted to the hospital with acute peritonitis secondary to ischemic perforation of the sigmoid colon. Angiography results revealed thrombosis of the superior mesenteric artery. LA and ANA were detected in the blood serum. The diagnosis of CAPS was based on fulminant damage to four organs: intestines, lungs, kidneys, and spleen. After successful therapy, she was discharged from the hospital, but 2 months later the patient was diagnosed with SLE due to ongoing circulation of LA.
4. Diagnostic Difficulties
Due to the variety of CAPS clinical manifestations, it can be difficult to diagnose. Huang et al. [
58] suggested using a history of arterial hypertension, anemia (OR 116.231, 95% CI 10.512–1285.142), elevated LDH levels (OR 59.743, 95% CI 7.439–479.815), and proteinuria (OR 11.265, 95% CI 2.118–59.930) in laboratory test results as an early predictor model in patients with an established diagnosis of APS.
Ferritin is an important signaling molecule and a direct mediator of the immune system. There is a close association between hyperferritinemia and such severe conditions as CAPS, macrophage activation syndrome, and septic shock. In a study by Rosário et al. [
59], 71% of patients with catastrophic APS had high serum ferritin levels (more than 1000 ng/mL).
If a patient has positive APLA titers, the diagnosis of APS cannot always be established. According to modern data, APLAs are present in the plasma of 1–5.6% of healthy people, and the antibody titer increases with age [
60]. The triggering mechanisms and causes of the presence of APLA have not yet been studied, as well as the full range of possible clinical manifestations. The presence of APLA may be a sign of inflammation or cancer, but further development of thrombosis does not always occur [
61,
62].
Sepsis is an extremely dangerous condition that develops in response to an infection.
The presence of two or more of the following symptoms of SIRS is necessary to confirm sepsis.
- (1)
-
Body temperature more than 38 °C or less than 36 °C;
- (2)
-
Pulse rate more than 90/min;
- (3)
-
Respiratory rate more than 20/min or PaCO2 less than 32 mmHg (4.3 kPa);
- (4)
-
Laboratory tests: white blood cell count more than 12,000/mL or less than 4000/mL or more than 10% immature cells [
63].
Distinguishing sepsis from CAPS is not always easy [
64,
65]. Tucker et al. [
66] presented a case report of a patient diagnosed with probable CAPS who developed inflammatory bowel disease with bloody diarrhea and abdominal pain. The patient’s medical history indicated a history of spontaneous abortions.
Heparin-induced thrombocytopenia (HIT) is an immune form of thrombocytopenia caused by unfractionated heparin (UFH) or, rarely, low molecular weight heparin (LMWH) use. In HIT, the formation of the heparin–platelet factor 4 (PF4) immune complex leads to the activation of platelets, monocytes, and endothelial cells and the release of tissue factor [
68,
69].
As a result, blood coagulation is hyperactivated and the risk of both arterial and venous thrombosis increases [
70]. Immune complexes (heparin–PF4) can also be detected in APLA-positive patients who have not taken heparin, due to the formation of autoantibodies to PF4.
CAPS is a thrombotic microangiopathic syndrome (TMA) and requires differential diagnosis in various urgent conditions. TMA includes heterogeneous clinical conditions characterized by arteriolar and capillary thrombosis with the development of Coombs-negative hemolytic anemia, thrombocytopenia, and ischemic organ damage resulting from microvascular occlusion by thrombi up to multiple organ failure.
CAPS in pregnancy creates many diagnostic challenges due to its wide range of clinical manifestations and similarities with other obstetric complications and microangiopathic diseases, such as hemolytic uremic syndrome (HUS), HELLP syndrome, thrombotic thrombocytopenic purpura (TTP), severe pre-eclampsia, DIC, septic shock, and SIRS [
71,
72]. Skin necrosis is one of the most striking clinical symptoms of CAPS.
5. Principles of Treatment
Due to the severity of clinical manifestations and high mortality in CAPS, the choice of adequate therapy is extremely important.
The primary goal of therapy should be to suppress thromboinflammation by reducing excessive complement activation, cytokine levels, NET formation, and excessive thrombin generation. Treatment should not be delayed unless all CAPS criteria are met, especially if there is a history of APS. If CAPS is suspected, aggressive therapy should be initiated immediately. Because CAPS occurs in only 1% of patients with APS, recommendations are based on case reports and expert opinion. An analysis of more than 500 cases of the CAPS registry [
35] formed the basis for first-line therapy for CAPS, including:
- (1)
-
Trigger elimination (termination of pregnancy, treatment of infection, etc.) if possible;
- (2)
-
Full anticoagulation with LMWH at a dose of 1 mg/kg (in the case of enoxaparin) every twelve hours;
- (3)
-
Glucocorticoids;
- (4)
-
Plasmapheresis with plasma exchange.
This approach reduced mortality by 37% and increased the recovery rate by 78% [
35].
IVIG therapy in addition to triple therapy is included in the international consensus guidelines for CAPS. There are no absolute recommendations regarding the dose or duration of IVIG administration, but it is generally recommended after the last day of plasmapheresis. Two common regimens have been proposed: 2 g/kg body weight for 2–5 days or 400 mg/kg for 5 days [
80].
6. Prognosis and Prevention
6.1. Prognosis
Due to the increasing number of new clinical cases of patients with CAPS, the European APLA Forum has established an international registry of patients with CAPS (CAPS Registry), which currently includes approximately 600 patients. High mortality of patients with CAPS is associated with SLE, lung and kidney damage, and also depends on the age of the patients (over 36 years old) and the APLA titer [
6]. Other long-term manifestations of CAPS include thrombosis and microantigopathic hemolytic anemia. There are not many cases of relapse of CAPS—18 episodes of “relapse” in eight patients were recorded.
6.2. Prevention
CAPS is a life-threatening condition that, despite treatment, is still associated with high mortality among patients and requires the development of effective preventive measures. These include identifying patients with risk factors: the presence of a high-risk APLA profile, t-APS and/or o-APS with high APLA titers, perinatal or perioperative withdrawal of anticoagulants, and infection. Prevention of CAPS requires anticoagulant prophylaxis before and after surgical interventions, including even trivial manipulations (biopsy, etc.), even in patients with o-APS and low antibody titers, as well as earlier genetic testing for thrombophilia [
9]. The use of LMWH in such cases has advantages not only because of its anticoagulant but also anti-inflammatory properties. Prevention and treatment of infection and timely delivery in high-risk groups of pregnant women also seem warranted.
6.3. CAPS and SARS-CoV-2
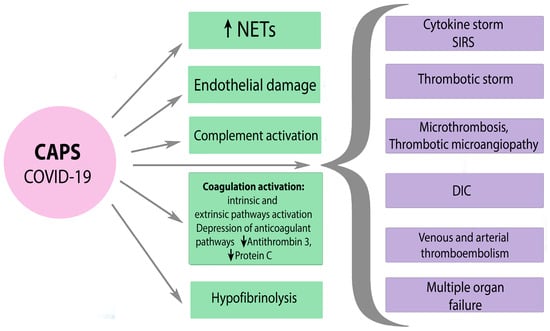
A severe SARS-CoV-2 infection is characterized by an hyperinflammatory reaction, endothelial dysfunction and systemic microthrombosis followed by multiple organ failure, increased ferritin, and other indicators, which are associated with high mortality among patients [
100]. Clinical manifestation of thrombophilia that occurs during severe SARS-CoV-2 infection is similar to the symptoms of CAPS. SARS-CoV-2 can lead to the development of CAPS in APS-positive patients due to indirect activation of complement and the associated procoagulant state [
101]. The mechanisms of hemostasis disorders in severe SARS-CoV-2 infection and CAPS are diverse and similar at the same time. There is often simultaneous activation of many pathways, including activation of platelets, external and internal pathways of coagulation cascade, suppression of fibrinolysis, etc. with the development of DIC, thrombotic microangiopathy, and even local intravascular coagulation in the lungs. Poorly controlled inflammation and hyperactivation of blood clotting cause not only a cytokine storm but also a thrombotic storm in severe COVID-19 and CAPS (
Figure 4).
Figure 4. The mechanisms of hemostasis disorders in severe SARS-CoV-2 and CAPS. CAPS—catastrophic APS, NETs—neutrophil extracellular traps, SIRS—systemic inflammatory response syndrome, DIC—disseminated intravascular coagulation. Black arrow down—decrease; black arrow up—increase.
7. Conclusions
CAPS is a multifactorial disease that requires an interdisciplinary approach and highly qualified medical care, adequate and timely diagnosis, and appropriate prevention. The variety of clinical symptoms often leads to late diagnosis of this condition with high mortality. Further study of the pathological mechanisms of CAPS and the role of thromboinflammation can significantly expand the prevention and treatment of this life-threatening condition through the use of capable drugs that can interrupt the crucial pathological pathways.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25010668
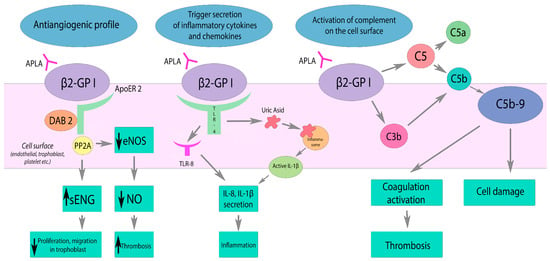 Figure 1. Effects of APLA on the complement system, inflammation, and vascular tone. β2GP1—β2-glycoprotein I, ApoER2—apolipoprotein E2 receptor, DAB2—Disabled-2, PP2A—Protein phosphatase 2A, eNOS—nitric oxide synthase, sENG—soluble Endoglin, TLR-4—toll-like receptor 4, TLT-8—toll-like receptor 8. APLA reduces eNOS activity via ApoER2 interaction. Decreased NO production results in impaired vasodilatation and endothelial dysfunction. APLA activates TLR and inflammasome pathways, triggering the secretion of inflammatory cytokines and chemokines. APLA activates complement on the cell surface, leading to the coagulation activation and cell damage by C5b-9 deposition. Black arrow down—decrease; black arrow up—increase.
Figure 1. Effects of APLA on the complement system, inflammation, and vascular tone. β2GP1—β2-glycoprotein I, ApoER2—apolipoprotein E2 receptor, DAB2—Disabled-2, PP2A—Protein phosphatase 2A, eNOS—nitric oxide synthase, sENG—soluble Endoglin, TLR-4—toll-like receptor 4, TLT-8—toll-like receptor 8. APLA reduces eNOS activity via ApoER2 interaction. Decreased NO production results in impaired vasodilatation and endothelial dysfunction. APLA activates TLR and inflammasome pathways, triggering the secretion of inflammatory cytokines and chemokines. APLA activates complement on the cell surface, leading to the coagulation activation and cell damage by C5b-9 deposition. Black arrow down—decrease; black arrow up—increase.