Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Among the indole-based oximes and oxime ethers, derivatives of indirubin (5) have been extensively investigated for their anticancer activity.
- oximes
- oxime ethers
- anticancer activities
1. Introduction
Cancer is a generic term used to describe a broad spectrum of diseases characterized by the rapid formation of abnormal cells that grow uncontrollably, extending beyond their usual boundaries and invading adjacent parts of the body while destroying normal tissues [1,2,3]. This condition can affect any part of the body, with the 2020 estimates from the World Health Organization (WHO) indicating that the most common types in terms of new cases were breast cancer (2.26 million cases), lung cancer (2.21 million cases), colon and rectal cancer (1.93 million cases), prostate cancer (1.41 million cases), nonmelanoma skin cancer (1.20 million cases), and stomach cancer (1.09 million cases), with the total number of new cases estimated to 19.3 million [4,5,6,7]. In fact, as mortality rates for other leading causes of death such as stroke and coronary heart disease decline in many parts of the world, cancer is emerging as the deadliest disease worldwide, and according to the WHO estimates, cancer ranks as the first or second leading cause of death before the age of 70 years in 112 out of 183 countries, and as the third or fourth leading cause in about 23 other countries [4,5,6,7,8,9]. In 2020, cancer was responsible for nearly one in six deaths, accounting for nearly 10 million deaths globally [4,5,6,7]. Compounding the issue, the overall cancer hardship across the globe is expected to reach 28.4 million cases by 2040, representing a 47% increase from 2020 [4,10,11]. One unique characteristic of cancer, unlike many other diseases, is its higher overall incidence in developed countries compared to transitioning or low-income countries, with rates ranging from 2 to 3 times higher [4,9,11,12]. This situation is further worsened by the prevailing trend of resistance to numerous available therapies, added to their pervasive side effects [13,14,15,16,17].
Fortunately, novel approaches to cancer treatment through a surgical targeting of tumors’ microenvironment, such as longitudinal single-cell profiling of chemotherapy [18,19] or targeted immunotherapies, including chimeric antigen receptor (CAR) cell therapy [20,21], immune checkpoint inhibitors [22,23,24], monoclonal antibodies [25,26,27], and immune system modulators [28,29,30], are demonstrating significant promise across various types of cancers. Nevertheless, small molecules have always been the bedrock of cancer therapy and are predicted to continue playing a critical role in the future. One such class of small molecules is oximes.
Oximes are nitrogen-containing molecules from the imine family, characterized by the general formula (RR′C=N–OH). They are available in the form of aldoximes or ketoximes, and have been well-known since the 1960s to be widely distributed across various species in all realms of life [31,32], with the first synthetic member of the family (methylglyoxime, 1) reported in 1882 by Meyer and Janny [33]. In plants, they are viewed as critical metabolic bifurcation points between general and specialized pathways, with the majority of plant oximes originating from amino acids through processes catalyzed by the cytochrome P450 family of enzymes [31,32]. As a result, the structures of naturally occurring oximes are not very complex, as they mirror their parent amino acids. This can be observed with the first oximes from plant origin, namely isobutyraldoxime (2) [34] (derived from valine) and phenylacetaldoxime (3) [35] (derived from phenylalanine), which were identified in 1967 from Linum usitatissimum and Tropaeolum majus, respectively (Figure 1). Additionally, indole-3-acetaldehyde oxime (4) (derived from L-tryptophan) was isolated from Brassica oleracea just a year later [36]. In fact, plant metabolites such as auxin, cyanogenic glucosides, glucosinolates, and other bioactive volatile compounds were also shown to originate from oximes [32,36,37]. This family of compounds and their derivatives has also been suggested to play key roles in growth regulation, plant defense, pollinator attraction, and plant communication with the surrounding environment [38,39,40]. Furthermore, compounds derived from oximes can serve as quenchers for reactive oxygen species in plants or act as storage compounds for reduced nitrogen, which may be released on demand through the activation of endogenous turnover pathways [31,32,41].
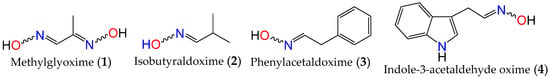
Figure 1. First synthetic and earliest amino acid-derived oximes from plants.
2. Anticancer Activity of Indole Based-Oximes and Oxime Ethers
Among the indole-based oximes and oxime ethers, derivatives of indirubin (5) have been extensively investigated for their anticancer activity. This purplish-red bisindole alkaloid known as the key active ingredient in traditional Chinese medicine herbal formulas, has been effective in treating human acute promyelocytic leukemia (APL) [54,55,56,57]. In fact, this Chinese concoction composed of Radix psudostellariae and Salvia miltiorrhiza, has demonstrated compelling synergistic effects in inducing differentiation of acute promyelocytic leukemia cells in vitro. Additionally, it has shown significant therapeutic activity in murine acute promyelocytic leukemia animal models [58,59] and thus has advanced into clinical trials for treating childhood acute promyeloid leukemia in China [55,57].
Numerous studies have also indicated that indirubin possesses a strong affinity for cyclin-dependent kinases (CDKs), which play an essential role in controlling cell cycle and proliferation [60,61]. This molecule was also shown to interact with glycogen synthase kinase-3β (GSK-3β) by interfering with the ATP-binding site [62], with its anticancer activity arising through the induction of cell cycle arrest in the G1 or G2 phase of tumor progression, leading to apoptosis in various cancer cell types [60,63,64]. While CDKs are key enzymes governing cell cycle progression [65], GSK-3β has been reported to play important roles in transcription by regulating the activities of key factors, such as NFκB [66], transcriptional factor EB [67], and in cell signaling pathways, such as growth factor signaling [68] and Wnt signaling [69].
However, indirubin’s therapeutic effectiveness is hindered by its poor water solubility, inadequate pharmacokinetic properties, low absorption rate, and significant gastrointestinal toxicity [55,70]. These factors pose substantial obstacles to its clinical application. Consequently, multiple strategies have been employed to enhance the pharmacodynamic and pharmacokinetic properties of this family of compounds. One such strategy involves transforming the carbonyls from the indirubin’s scaffold into oxime or oxime ether pharmacophores, as illustrated in Figure 2. This approach has led to the preparation of a wide variety of indirubin C-3′-oxime (6) derivatives, many of which exhibited superior properties compared to the naturally occurring parent alkaloid, including enhanced water solubility and better selectivity [55,61,70,71,72]. For example, while exploring the anticancer activity of a series of N1-alkylindirubin C-3′-oximes against MCF-7, LOVO, and LNCAP cells, Wang et al. [73] showed that many of these derivatives exhibited moderate cytotoxicity, with 7 being the most active across all these cell lines, displaying IC50 values ranging from 6.72 to 13.38 μM.
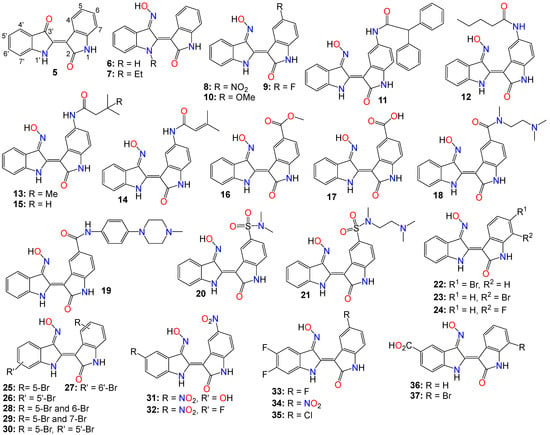
Figure 2. Illustrative examples of anticancer indirubin-3′-oxime derivatives, bearing a diversity of substitution patterns on the core bis-indole scaffold.
The effects of various substitution patterns on the activity of these compounds have also been extensively investigated. In fact, Lee et al. [74] demonstrated that indirubin-5-nitro-3′-oxime (8) exhibits superior antiproliferative activity against A549, HT-1080, and HL-60 cells compared to indirubin-3′-oxime, with IC50 values of 5.4, 5.9, and 9.2 µM, respectively. Further studies have indicated that the presence of an electron-withdrawing group at the 5-position enhances the anticancer activity of these compounds. Out of the 29 molecules synthesized and evaluated for their effects on MV4-11 cells (with FLT3/ITD mutation), compound 9 displayed the highest inhibition potency, with an impressive IC50 value of 0.072 μM [75]. Moreover, this molecule was found to selectively inhibit the activity of FLT3 kinase and induce cell cycle arrest at the G0/G1 phase in MV4-11 cells [75]. Additional structure–activity relationship studies revealed that a sulfonate group at the 5-position improved the inhibitory activity against FLT3 kinase. However, this substitution was found to be detrimental to antiproliferative activity due to the resulting molecule’s low cell membrane permeability [75]. Nevertheless, 5-methoxyindirubin 3′-oxime (10) has been demonstrated to inhibit PDAC cell proliferation, with the administration of compound 10 leading to the inhibition of PDAC xenograft growth in BALB/c nu/nu mice, and further studies suggesting that this analog might exert its activity by inducing G2/M phase arrest in PDAC cells through the inhibition of CDK1/cyclin B1 levels, subsequently leading to apoptosis [76]. Furthermore, 5-diphenylacetamido-indirubin-3′-oxime (11), a mitochondria-targeting antileukemic agent, exhibited strong inhibitory activity against a panel of drug-sensitive and drug-resistant primary and malignant leukemia cell lines [77]. This compound rapidly collapses the mitochondrial membrane potential (MMP), followed by the release of cytochrome c into the cytosol, and a severe depletion of cellular ATP [77]. These findings indicate that this molecule induces leukemia cell death through a mechanism distinct from that of many other indirubin analogs [77].
Death-associated protein (DAP) kinases, including DRAK2 (apoptosis-inducing kinase 2), are well known to perform apoptosis-related tasks in cells [78]. A number of studies have shown that DRAK2 is engaged in regulating the activation of T lymphocytes as well as pancreatic β-cell apoptosis in type I diabetes [78]. This kinase also plays an important role in the development of tumor-related diseases via diverse mechanisms, and has been shown to be involved in the exacerbation of alcoholic fatty liver disease (NAFLD) through SRSF6-associated RNA selective splicing, the regulation of chronic lymphocytic leukemia and acute myeloid leukemia, as well as the progression of colorectal cancer [78].
A number of 5-amido-indirubin-3′-oxime derivatives have been shown to exhibit potent inhibitory activity against DRAK1 and DRAK2 [79], with further structure–activity relationship studies indicating that small aliphatic amide at the 5-position were better promotor of the inhibitory activity against DRAK2. Among them, 12 (IC50 3 nM and 51 nM), 13 (IC50 14 nM and 24 nM), 14 (IC50 14 nM and 120 nM), and 15 (IC50 8 nM and 21 nM) displayed strong inhibitory activity against DRAK1 and DRAK2, respectively [79], with the molecular docking study revealing that compound 12 binds to the ATP-binding site in DRAK2 by forming three key H-bond interactions, specifically with Glu111, Ala113, and Glu117 [79].
Indirubin-3′-oxime analogs with carboxylic acid, ester, carbamide or sulfonamide at the 5-position have also been studied for their anticancer activity. In fact, a 5-methyl acetate analog of indirubin-3′-oxime (16) demonstrated potent inhibition of FLT3 kinase activity in vitro, with an IC50 of 7.89 nM [80]. In contrast, the corresponding free carboxylic acid (17) analog showed relatively weak activity against FLT3 (IC50 of 3.19 μM) [80]. Interestingly, only the 5-methyl acetate analog exhibited a strong cytotoxic effect against MV4-11 cells, while the carboxylic acid analog was inactive. Further studies revealed that compound 16 inhibited the phosphorylation of downstream STAT5, promoted PARP cleavage in MV4-11 cells, and induced apoptosis and cell cycle arrest in the G1 phase [80]. On the other hand, Merz and coworkers designed and synthesized a series of indirubin-5-carboxamide oxime derivatives with improved water solubility. These compounds were evaluated for their antiproliferative activity against LXFL529L cells, with 18 and 19 exhibiting noticeable inhibitory activity [81].
While evaluating the cytotoxicity of a panel of 5-sulfonamide indirubin oxime analogs with enhanced water solubility and improved physicochemical and pharmacological properties, Jautelat et al. [82] indicated that many of these analogs possess potent inhibitory activity toward CDK2. These molecules also exhibited significant antitumor activity against MCF-7 cells, with compounds 20 (IC50 0.04 and 0.4 μM) and 21 (IC50 0.04 and 0.1 μM) being the most active in both CDK2 inhibitory activity assay and tumor growth inhibition assay against MCF-7 cells [82].
The effects of other substitution patterns on the anticancer activity of this family of compounds have also been explored. 6-bromoindirubin-3′-oxime (22) was shown to inhibit the activity of glycogen synthase kinase 3β (GSK3β) [83]. This compound suppresses the proliferation, invasion, and migration of ovarian cancer cells (A2780 and OVCAR3), reduces lamellipodia formation, and induces G1 arrest of the cell cycle [83]. Exposure of these cells to compound 22 led to a significant downregulation of mRNA and protein expression of cyclin D1 and MMP9 compared with untreated control cells. Moreover, 6-bromoindirubin-3′-oxime strongly reduces the formation of lung metastasis in the well-established 4T1 mouse model of aggressive breast cancer [83]. Subtoxic concentrations of this analog affect a number of key characteristics of the metastatic process, including the inhibition of adhesion, migration, and invasion of a variety of metastatic cell types in vitro [83]. Surprisingly, RNAi-mediated silencing of glycogen synthase kinase 3β and phosphoinositide-dependent protein kinase 1 (PDK1), both modulators of cellular metastasis targeted by 6-bromoindirubin-3′-oxime, did not affect invasive migration in this study [83]. Instead, the Jak/STAT3 signaling pathway appeared to play a major role by modulating downstream migration regulators such as C-terminal tensin-like protein and matrix metalloproteinase 2 [83]. However, PDK1 and GSK3β still contributed to the overall response to 6-bromoindirubin-3′-oxime, as silencing all three pathways resulted in almost a complete inhibition of migration, mimicking the response of compound 22 [83].
In contrast to its 5-bromo and 6-bromo isomers, 7-bromoindirubin-3′-oxime (23) exhibited only marginal inhibitory activity towards CDKs and GSK-3 [71,84,85]. However, this latter analog induced the appearance of large pyknotic nuclei, without exhibiting classical features of apoptosis such as chromatin condensation and nuclear fragmentation. The induced cell death was not accompanied by cytochrome c release or any measurable effector caspase activation [71,84]. This result indicates that, unlike other indirubin analogs, 7-bromoindirubin-3′-oxime triggers the activation of a nonapoptotic cell death pathway, possibly through necroptosis or autophagy. In this study however, the 7-fluoro derivative (24) appeared to have better inhibitory activity towards CDKs and GSK-3 than its 7-bromo counterpart [71,84]. Nevertheless, 7-bromoindirubin-3′-oxime significantly reduced cell viability in 14 thyroid carcinoma cell lines, with treated cells showing DNA fragmentation, cell cycle arrest, and lactate dehydrogenase release, but no LC3B cleavage [85]. In other words, this compound induced a nonclassical kind of cell death that was caspase-independent and did not involve DNA fragmentation [85].
A number of studies focusing on the investigation of the anticancer activities of indirubin-3′-oxime bearing a diversity of substitution patterns on the bisindole scaffold have also been reported. In a comparative analysis against indirubin-3′-oxime parent compound, Ichimaru et al. [86,87] prepared and evaluated a series of analogs with a variety of bromo- or dibromo-substituted patterns as illustrated by compounds 25–30 in Figure 2. This variation in substitution patterns did not appear to significantly affect the activity of the resulting compounds, with IC50 values ranging from 4.4 to 14 μM [86,87].
While exploring other substitution patterns, Kim and coworkers demonstrated the 5,5′-substituted derivatives of indirubin-3′-oxime are potent CDK inhibitors, as many of these compounds displayed potent inhibitory effects on CDK2, with over 90% inhibition potency at 1 μM concentration [88,89,90]. 5′-Hydroxy-5-nitro-indirubin oxime (31) and 5′-fluoro-5-nitro-indirubin oxime (32) not only displayed selective and significant inhibitory activities towards CDK1 and CDK2, but also exhibited good anticancer activity against several cancer cell lines [88,90]. Compound 31 was able to inhibit the phosphorylation of the retinoblastoma protein (Rb), which is a major substrate of CDK, as well as CDK2/cyclin E activity (IC50 = 2.16 nM) more efficiently when compared to its effect on the CDK1/cyclin B activity (IC50 = 13.8 nM) [75,89,90]. Docking studies indicated that compound 31 formed a new hydrogen between the 5′-hydroxy and Asp86 in the solvent-accessible region of CDK2, with the 3′-oxime moiety creating a hydrogen bond with Ile10 instead of Gln131 [75,88]. Further SAR studies revealed that small substituents at the 5′-position, such as -OH or -F, as well as electron withdrawing substituents at the 5-position, such as -NO2, significantly increased the inhibition potency against CDK2 [75,88,90].
Compounds 31 and 32 were able to induce apoptosis in Imatinib-resistant chronic myeloid leukemia cells, as well as efficiently decrease the viability of CML-derived drug-sensitive and Imatinib-resistant K562 cells, both in vitro and in vivo [90]. Additionally, Park and co-workers indicated that compound 31 significantly decreased the viability of leukemia K562 cells with an IC50 value of 669 nM, while also inhibiting Imatinib-resistant K562R cells with a similar IC50 value of 783 nM [90]. A combination of 31 and Imatinib also appeared to increase the number of apoptotic K562R cells compared to the effect of either 31 or Imatinib, with the former (31, 15 mg/kg, but not Imatinib) significantly inhibiting the growth of K562R tumor cells in vivo [88,90,91].
Using the knowledge gathered from the elucidation of the X-ray structure of 5-bromoindirubin and CDK2, Yan et al. [92], prepared various 5′,6′-difluoro-indirubin derivatives, among which compounds 33–35 showed potent inhibitory effects on both CDK2/cyclin E1 and CDK9/cyclin T1 at nanomolar or low micromolar levels. Docking studies indicated that compound 35 positions well into the ATP pocket and exhibited similar bindings with hinge region residues in CDK9 and CDK2, with this 5-chloro analog unexpectedly forming two additional halogen bonds with Lys48 and Asp167 residues in CDK9, whereas, such a halogen bond was absent in CDK2, indicating that the selectivity exhibited by 35 towards CDK9 might be related to the formation of these halogen bonds. Yet, while compounds 33 and 34 showed a significant cytotoxic activity against five human cancer cell lines with IC50 values at low micromolar levels, 35 displayed only a marginal activity, a situation that might be attributable to its poor solubility [92].
Dual-specificity tyrosine phosphorylation-regulated kinases (DYRKs) are well known to be involved in multiple cellular functions, including intracellular signaling, mRNA splicing, chromatin transcription, DNA damage repair, cell survival, cell cycle control, differentiation, endocytosis, and synaptic plasticity [93], to name just a few. Myrianthopoulos et al. reported the synthesis of indirubin-3′-oxime-based selective DYRK inhibitors [94]. They revealed that a carboxylate substitution at the 5′-position in combination with a bromine at the 7-position significantly promoted selective DYRK inhibition, as illustrated with compounds 36 (IC50 0.31 and 0.35 μM) and 37 (IC50 0.21 and 0.13 μM) [94].
To rationalize the observed affinity and selectivity profiles, docking studies of compound 37 to a DYRK1a-derived homology model of DYRK2 revealed the existence of two distinct binding modes characterized by a 180° flip of the indirubin core in relation to either its primary or secondary axis [94]. In both cases, the typical hydrogen bond triplet between the indirubin pharmacophore and the kinase hinge was either disrupted (mode I) or inverted (mode II), with the 5′-carboxylate forming a salt bridge either with Lys165 (mode I) or with the catalytic Lys178 (mode II) [94]. This binding mode appears to be different from the experimental indirubin-kinase binding established in previous studies [95].
The crystal structure of 37 bound to DYRK2 (Figure 3) at a resolution of 2.28 Å confirmed that the binding mode was indeed inverted in comparison to previously reported indirubin-kinase complexes geometric arrangement [95], and consistent with the predicted binding mode II [94]. In fact, not only the crystal structure revealed that the predicted salt bridge anchoring the anionic 5′-carboxylate to the sidechain of Lys178, but also disclosed an additional hydrogen bond between the 5′-carboxylate and the NH backbone of Asp295, with three water molecules participating in the hydrogen bond network by bridging the inhibitor carboxylate and Lys178 ammonium groups to the sidechains of Asp295 and Glu193 [94].
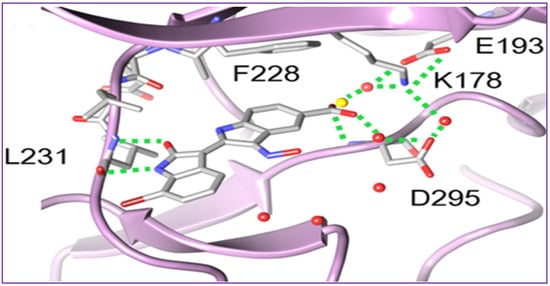
Figure 3. Crystal structure of the 37-DYRK2 complex revealing a nontypical binding mode, in which the orientation of the indirubin core is in good accordance with the predicted binding mode.
Despite the numerous substitutions performed on the core bisindole skeleton, indirubin C-3′-oxime derivatives still exhibited solubility issues, particularly in vivo within live tissue. These poor solubility and inadequate pharmacokinetic properties are primarily attributed to the high polarity and hydrophilic nature of the oxime functional group. To modulate the hydrophilic/hydrophobic balance of this family of compounds, various substituents, including glycosides, polyhydric alcohols, epoxides, amide chains or their corresponding salts, were added to the 3′-oxime functional group, effectively transforming them into an oxime ether.
As such, Nam et al. [96] synthesized several indirubin-3′-oxime ethers by introducing diverse polyether or polyhydric alcohol chains to the hydroxyl group of the 3′-oxime. They demonstrated that compounds 38 and 39 strongly inhibited Stat3 signaling in four human prostate and breast cancer cell lines [96]. Additionally, 39 was found to directly inhibit c-Src kinase activity in vitro (IC50 = 0.43 μM) [96]. Treatment with the same compound appeared to suppress the tyrosyl phosphorylation of c-Src kinase’s downstream target protein Stat3, while concurrently downregulating the Stat3-associated proteins Mcl-1 and Survivin, triggering MDA-MB-468 breast cancer cell apoptosis [96].
Owing to the fact that many epoxides can be further metabolized by epoxide hydrolase in the cellular metabolic environment to generate the corresponding polyhydric alcohols, Miyairi and co-workers prepared a series of indirubin-3′-(O-oxiran-2-ylmethyl)-oxime derivatives designed to enhance cell permeability, and thus serving as prodrugs [86,87]. The anticancer activity of each molecule was evaluated on HepG2 cells through a comparative analysis against their respective indirubin-3′-oxime parent compounds with appropriate substituents [76,86,87]. This study indicated that all indirubin 3′-(O-oxiran-2-yl)oxime derivatives consistently outperformed their corresponding indirubin-3′-oxime intermediates [86,87].
A docking study with 40 at the ATP-binding site of GSK-3β designed to evaluate the positioning of this molecule within the site, including the interactions between the oxirane and amino acid residues, suggested that the oxirane group of 40 is positioned closely enough to Cys-199 to form a covalent linkage between them [86,87]. It should be mentioned that Cys-199 is a critical amino acid for GSK-3β kinase activity [62]. Given that halogen atoms in organic compounds have exhibited strong affinity for the sulfur atoms in cysteine and methionine residues in a number of proteins [97], the participation of Cys-199 in enhancing binding affinity through sulfur-halogen interaction was anticipated. The authors concluded that the sulfur-halogen interaction forces the oxirane moiety close to Cys-199, thereby accelerating the covalent bond formation between the oxirane and the thiol [76,86,87]. In addition, Kurita et al. [98] demonstrated that these indirubin-3′-(O-oxiran-2-ylmethyl)-oxime derivatives possess potent cytotoxicity against human neuroblastoma IMR-32 and SK-N-SH cell lines, with IC50 values of 0.16 and 0.07 µM, respectively. These molecules induce caspase-independent apoptosis by inhibiting DNA repair and causing DNA fragmentation in IMR-32 cells [76,98].
While exploring the effect of different substitution patterns on the behavior of these analogs, Ichimaru et al. [86,87] found that many indirubin-3′-oxime derivatives were potential anticancer agents, with compounds 41–44 exhibiting significant inhibitory activity against HepG2 cells, with IC50 values of 0.62, 1.7, 1.3, and 1.6 μM, respectively [86]. Notably, compound 41 (with a 6-bromo substituent) displayed the most promising anticancer profile, whereas the activity of compounds 42 and 44 was not superior to that of their corresponding unsubstituted analog 40 [86,87,98].
Further investigations by other research groups have indicated that compound 39 possesses a broad range of in vitro and in vivo activities against numerous cancer cell lines. For instance, 39 was found to significantly inhibit the growth of CT-26 allografts in syngeneic BALB/c mice without affecting body weight [99,100]. Immunohistochemistry revealed that 39 could induce apoptosis and hinder tumor angiogenesis, leading to substantial impediment of cell proliferation, migration, and tube formation in vascular endothelial growth factor (VEGF)-treated HUVECs [99,100]. In vivo studies showed that this analog has the potential to inhibit the formation of subintestinal vessels in a zebrafish model in a dose-dependent manner [100]. In a different study, Nam et al. [101] further indicated that 39 significantly blocked tyrosyl phosphorylation of Stat5, while inhibiting Stat5 DNA-binding activity in human K562, KCL-22M, and primary CML cell lines, with treatments with 39 strongly decreasing the autophosphorylation of Src and SFKs in K562 and KCL-22M cells at 5 μM, and in primary CML cells. This compound also induced apoptosis in chronic myelogenous leukemia cells by downregulating the expression levels of Stat5 target proteins Mcl-1 and Bcl-xL, highlighting the potential of 39 to inhibit SFK/Stat5 signaling in leukemia cells [101]. During an investigation into the effects of different substitution patterns on the anticancer activity of compound 39, Cheng et al. [102] noted that compound 45, a 5-methoxy derivative of 39, could significantly block TGFb/BMP signaling by inducing ubiquitin-proteasome-mediated depletion of total R-Smads. This molecule also exhibited a strong inhibition of cell viability across several pancreatic cancer cell lines (Panc-1, MIA-PaCa2, BxPC3, and AsPC1), with IC50 values ranging from 0.76 to 2.2 μM [103].
Nevertheless, while further investigating the physicochemical and cell permeability properties of these molecules, Heshmati et al. [104] demonstrated that water solubility remained a significant hurdle for drug permeation, although a Calcein-AM uptake assay also indicated that 39 was not a good substrate for P-glycoprotein, suggesting that this compound could be a promising candidate for oral delivery due to its high membrane permeability [104]. It is worth mentioning that Jakobs et al. [64,105], while investigating some glycoside derivatives of indirubin C-3′-oxime, showed that 39, 46, and 47 exhibited potent cytotoxicity effects against LXFL529L, MCF-7, and HCT-116 cell lines at low micromolar concentrations [105], with compound 46 proven to block pRb phosphorylation in LXFL-529L cells [64].
Another strategy used to improve the physicochemical properties of this family of compounds is to attach a quaternary ammonium group to the oxime moiety. In line with this approach, Ginzinger et al. [106] prepared a series of carnitine oxime ether and choline oxime ether prodrugs. They not only investigated the activities of these compounds against three human cancer cell lines (A549, CH1, and SW480), but also assessed their suitability for clinical administration [106]. Compounds 48 (IC50 7.6, 1.8, and 5.6 μM), 49 (IC50 13, 1.3, and 2.5 μM), and 50 (IC50 1.0, 0.28, and 0.52 μM) exhibited low micromolar activity against A549, CH1, and SW480, respectively, with 50 being the most potent [106]. Furthermore, a 5-nitro derivative (51) of these analogs exhibited a significant anticancer activity against HCT-116 and MDA-MB-231 cell lines, with IC50 values of 0.302 and 0.738 μM, respectively [107]. This compound also significantly inhibited cell invasion and induced apoptosis in MDA-MB-231 cells, while blocking cell metastasis in a zebrafish human tumor xenograft model without any apparent toxicity [107]. Further assays indicated that compound 51 acts as a dual inhibitor of GSK-3β (IC50 5.46 nM) and Aurora A (IC50 0.22 μM) [107].
2-(N-methylpiperazin-1-yl)ethyl and 2-(N-methylpiperazinium)ethyl hydrochloride derivatives of these analogs, also exhibited some anticancer activity against LXFL and MCF7 cells in vitro, with compounds 52 (IC50 of 0.86 and 0.80 μM) and 53 (IC50 of 1.1 and 0.85 μM) displaying the highest potency [72]. Notably, compound 53 exhibited greater water solubility than its counterpart (52), and also exhibited selective inhibitory activity on IGF-1R, with an IC50 value of 169 nM [72]. This compound also induced apoptosis and caused cell cycle arrest, primarily at the G2/M phase in MCF-7 cells [72].
Jeong et al. [108,109,110] further demonstrated that these piperazinium hydrochloride derivatives of indirubin-3′-oxime were potent inhibitors of FLT3 kinase and MV4-11 cells, with compounds 53 and 54 exhibiting the highest potency. Docking studies suggested that compound 53 bound tightly to the ATP binding pocket in FLT3, surrounded by residues Leu616, Leu818, Cys828, and Tyr693, and forming approximately five hydrogen bonds with these residues [108,109]. While the indirubin core formed two significant hydrogen bonds with the backbone Cys694 residue, the tertiary amine of the piperazine moiety formed a hydrogen bond with Asn816, and the terminal amino group established two hydrogen bonds with both Asn816 and Asp829 [108,109]. Moreover, compound 53 effectively inhibited cell growth in vivo in an MV4-11 xenograft model following a daily oral administration of 20 mg/kg, without inducing significant hERG-related cardiotoxicity [108,109,110].
Conversely, compound 52 inhibited the growth of TT thyroid carcinoma cells by suppressing cell proliferation and inducing apoptosis [110]. This compound was also found to suppress the RET signaling pathway via a downregulation of the phosphorylation of RET kinase as well as the downstream Shc and ERK1/2 [108,109].
Dicationic piperazinium dihydrochloride derivatives of indirubin-3′-oxime have also been extensively studied for their anticancer activities. In fact, compound 55 demonstrated activity against Abl1 and T315I mutant Abl1 leukemia cells, with IC50 values of 0.87 and 9.4 μM, respectively [111]. This compound also exhibited inhibitory effects on both c-Src and Abl kinases, with SAR studies indicating that the presence of a bromine atom at the 6-position and a flexible alkylamino chain on the 3′-oxime functional group enhances cytotoxicity [111]. Conversely, the trifluoromethyl or bromine group at the 7-position in compound 56 seems to be detrimental to its cytotoxic activity [111]. However, this latter compound showed activity against SH-SY5Y, while displaying only marginal inhibitory activity towards CDKs and GSK-3α/β kinases [112]. Furthermore, compound 56 has been shown to significantly suppress the viability of four human melanoma cell lines (A2058, A375, G361, and MeWo) in vitro in a dose-dependent manner [113]. However, the 5-methyl acetate derivative (57) exhibited the highest potency among these analogs, displaying an excellent in vitro FLT3 inhibition, with an IC50 value of 3 nM, while blocking the phosphorylation of downstream STAT5 [110]. This same compound exhibited highly selective anticancer activity against leukemia MV-4-11 cells, with an IC50 value of 1.2 nM, achieved through G2/M phase arrest and apoptosis induction. Additionally, it inhibited c-Met kinase and blocked c-Met phosphorylation, along with downstream signaling pathways (Erk1/2, STAT3, STAT5, and Akt) in a dose-dependent manner [114]. In an in vivo study, this compound demonstrated a significant inhibitory activity in MV4-11 tumor xenograft model, without causing alterations in the mice’s body weight [110]. Further studies by Vougogiannopoulou et al. [115] indicated that compound 58, the 6-bromo derivative, exerted strong activity against neuroblastoma SH-SY5Y cells, while also significantly inhibiting GSK-3 activity, with an IC50 value of 1.3 nM.
Several indirubin C-3′-oxime ethers bearing a 2-(pyrrolidin-1-yl)ethyl substituent at the oxime functional group have also been shown to strongly inhibit the growth of a series of leukemia cells, while simultaneously blocking the activity of both c-Src and Abl kinases [111]. Among these compounds, 59 and 60 exhibited potent cytotoxicity against wild-type KCL-22 cells and T315I mutant KCL-22 cells in the sub-micromolar range [111]. Additionally, compounds 61 and 62 exhibited significant growth inhibition in SH-SY5Y, HCT116, F1, and Huh7 cell lines [112]. These compounds only displayed marginal inhibitory activity towards CDKs and GSK-3α/β kinases, suggesting that, unlike many indirubin derivatives, their anticancer activities might not be primarily mediated through a direct effect on CDK1, CDK5, and GSK-3α/β kinases [112].
Ultimately, Van et al. synthesized a number indirubin-3′-oxime ethers by attaching polyhydric alcohol chains with bulky 1,3,4-thiadiazole substituents to the hydroxyl of the oxime functional group [116]. Among these compounds, 63 and 64 exhibited only a moderate activity, albeit comparable to that of the parent indirubin-3′-oxime [116]. Further SAR studies indicated that the introduction of bulky substituents to the 3′-oxime group mostly resulted in a decrease in the anticancer activity of the resulting molecule [116]. The structure of all these indirubin-3′-oxime ether derivatives are summarized in Figure 4.
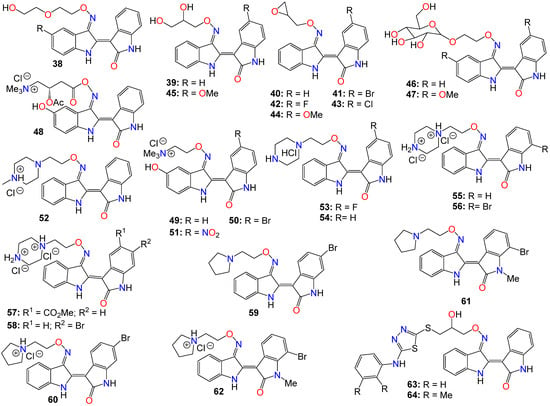
Figure 4. Illustrative examples of anticancer indirubin-3′-oxime ether derivatives.
While investigating the effect of different substituents at N1 of the bisindole core structure on the anticancer activity of the resulting indirubin-3′-oxime derivatives, Anh et al. [117] prepared a series of indirubin-based conjugates by attaching a range of hydroxyamides at that position, as illustrated in Figure 5. The inhibitory potential of the resulting molecules on the proliferation of cancer cell lines was investigated, with many of these conjugates exhibiting cytotoxic activity several magnitudes higher than that of the parent indirubin-3′-oximes [117]. Furthermore, compounds 65–67 significantly inhibited the proliferation of SW620, PC3, and NCI-H23 cells, with IC50 values in the sub-micromolar range. These compounds also displayed strong histone deacetylases (HDACs) inhibitory activity, with IC50 values as low as 22 nM. The inhibitory activity of 65 towards HDAC6 (IC50 0.007 μM) was 29-fold higher compared to its effect towards the HDAC2 isoform (IC50 0.205 μM) [117]. Docking studies revealed that 65 positioned into the HDAC6 pocket with the lowest energy and formed three hydrogen bonds with key residues, namely Tyr782, His610, and Asp649. However, it did not display any such hydrogen bond-type interaction within the HDAC2 pocket [117].
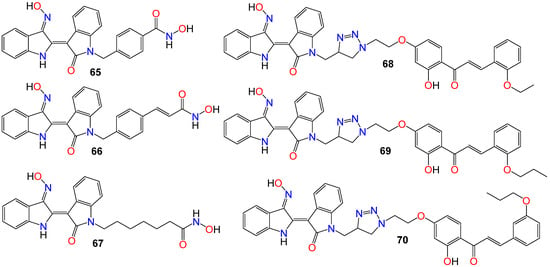
Figure 5. Illustrative examples of anticancer indirubin-3′-oxime derivative with substituents at N1 of the bisindole core.
The activity of a series of indirubin-3′-oxime conjugates, prepared by attaching different chalcone units at the N1-position of the core indirubin skeleton via 1,3,4-thiadiazole click chemistry, was also evaluated against four human cancer cell lines (HepG2, LU-1, SW480, and HL-60), with the human normal kidney cell line (HEK-293) used as a control [118]. Among them, compounds 68–70 displayed significant growth inhibitory properties on all cell lines, with the most potent compound 68 exhibiting IC50 values of 2.01, 1.30, 2.54, and 0.98 μM against HepG2, LU-1, SW480, and HL-60 cell lines, respectively [118]. Docking studies indicated that compound 68 forms three hydrogen bonds towards GSK-3β via two residues, namely Val135 and Thr138 [118]. Further SAR studies indicated that bulky and rigid substituents are not suitable for the 5′-position, as they led to reduced cytotoxicity and poor water solubility. In the NCI60 cell line panel, these compounds generally inhibited the growth of malignant tumor cells at low micromolar concentrations [118].
Other types of bisindole derivatives bearing an oxime moiety, not directly related to indirubin, have also undergone investigation for their anticancer activities. In fact, the antiproliferative activities of a series of 1′H,3H-[2,3′-biindol]-3-ones, designed and synthesized by Qu et al. [119], were explored for their inhibitory effects on seven human cancer cell lines, using HUVEC normal cells as a control. Initial structure–activity relationship (SAR) studies indicated that the presence of an oxime group at position 2 was crucial for the cytotoxic effects. Furthermore, the introduction of a polar substituent at C-5 and C-5′ significantly enhanced the anticancer activities of the resulting compounds [119]. Among these compounds, 71 exhibited the highest potency, with IC50 values of 6.09, 7.66, 9.92, and 4.31 μM against A529, MGC-803, HepG2, and T24 cell lines, respectively [119].
Subsequent investigations revealed that this compound induced apoptosis in T24 cells by elevating intracellular reactive oxygen species (ROS) levels and altering the balance of anti- and pro-apoptotic proteins, leading to mitochondrial dysfunction and activation of caspase-9 and caspase-3 [119]. Furthermore, both cell cycle analysis and Western blotting indicated that compound 71 effectively arrests the growth of T24 cells in the G1 stage, possibly influencing cell cycle regulatory proteins, particularly cyclin D1, while also causing a significant increase in the activity of p53, p21, and p16 [119].
While assessing a series of bis(indolyl)methane oximes against several tumoral cell lines, namely HepG2 (hepatocellular carcinoma), MDA-MB-468 (human breast carcinoma), RAW 264.7 (murine leukemic monocyte macrophages), THP1 (human acute monocytic leukemia), U937 (human leukemic monocytic lymphoma), and EL4 cells (murine T-lymphoma), Grosso et al. [120] demonstrated that compounds 72 and 73 exhibited potent cytotoxic effects across all tested cells. The IC50 values varied between 1.62 (THP1) and 23.9 μM (RAW) for 72, and between 10.7 (MDA) and 34.1 μM (U937) for 73. Notably, compound 72 displayed pronounced activity against nonadherent cell lines, with IC50 values ranging from 1.62 in THP1 to 1.65 μM in EL4 [120]. In contrast, 74 exhibited a considerably lower cytotoxicity in tumoral cell lines compared to the other two compounds, with IC50 values ranging from 35.7 (HepG2) to 124 μM (THP1), without displaying any selectivity. The difference in activity between the N-unsubstituted 72 and the N-methyl substituted derivative 74 was quite significant, underscoring the potential importance of a free amine interaction within the binding pocket through hydrogen bonds [120]. Conversely, the notably lower IC50 values observed for 72 in nonadherent cell lines in comparison to those obtained for 73 demonstrated that the presence of a bromine substituent contributes to higher cytotoxic activity. Unfortunately, these compounds exhibited only a marginal selectivity, with only about one to two-fold difference compared to their activity on nontumoral cells [120].
Dandu et al. [121] prepared and screened a series of dihydroindazolo[5,4-a]pyrrolo[3,4-c]carbazole oximes against recombinant human VEGF-R2 and TIE-2 receptor tyrosine kinases. Compounds 75, 76, and 77 exhibited activity against both TIE-2 (with IC50 values of 30, 25, and 26 nM, respectively) and VEGF-R2 (with IC50 values of 7, 4, and 4 nM, respectively), while also demonstrating favorable pharmacokinetic properties in rats [121].
Homology modeling and docking experiments revealed that 75 binds effectively within the ATP pocket of the TIE-2 model, with the lactam NH/CO moiety forming a bidentate donor/acceptor interaction with Glu903/Ala905 at the hinge region, with the anti-oxime orientation serving as an acceptor for the Asp982 backbone amide [121]. The hydrophobic cavity that accommodates the O-alkyl oxime is defined by Leu888, Leu976, Phe983, Gly984, Leu985, and Ile902. Indeed, a single-crystal structure solved for 75, confirmed both the trans-oxime orientation and the regiochemistry of the lactam carbonyl, as well as the orientation of the indazole N2 -methyl group [121].
A number of indeno[1,2-b]quinoxaline derivatives were also synthesized and evaluated for their antiproliferative effects. Among them, compounds 78 (IC50 0.87, 5.78, 0.82, and 0.64 μM), 79 (IC50 0.67, 6.88, 0.89, and 0.83 μM), 80 (IC50 0.45, 5.69, 0.68, and 0.82 μM), 81 (IC50 0.90, 0.81, 0.78, and 0.75 μM), 82 (IC50 0.68, 0.92, 0.78, and 0.32 μM), and 83 (IC50 0.66, 0.83, 0.91, and 0.63 μM) demonstrated the ability to inhibit the growth of MDA-MB231, H1299, PC-3, and Huh-7 cancer cell lines, respectively [122]. Compound 78 was found to be inactive against the growth of the normal human fetal lung fibroblast cell line (MRC-5), with an IC50 value of 31.51 μM. This study also demonstrated that compounds 78–80 (Figure 6) exhibited comparable inhibitory activities against topoisomerase I and topoisomerase II [122]. Mechanistic studies indicated that compound 78 induced cell cycle arrest at the S phase through the activation of caspase-3, -7, an increase in the protein expression of Bad and Bax, and a decrease in the expression of Bcl-2 and PARP, ultimately resulting in cell death [122]. Furthermore, compound 78 attenuated the levels of phosphorylated Src, Akt-1, and Akt-2 proteins, without affecting the total protein expression of Akt. Additional studies involving human hepatocellular carcinoma cells in a zebrafish xenograft assay confirmed the antitumor effect of 78 in vivo [122].A series of diarylmethyloxime derivatives containing 5-indolyl moieties were also found to be potent inhibitors of tubulin polymerization [123]. Their inhibitory activity was assessed against several cancer cell lines, including HeLa human cervix epithelioid carcinoma, A-549 human lung carcinoma, HL-60 human leukemic, and HT-29 human colon adenocarcinoma. Among these compounds, 84 exhibited the highest activity against HeLa, HL-60, and HT-29 cell lines, with IC50 values of 0.36, 0.33, and 0.11 μM, respectively. However, this compound showed no activity against the A-549 cell line [123].
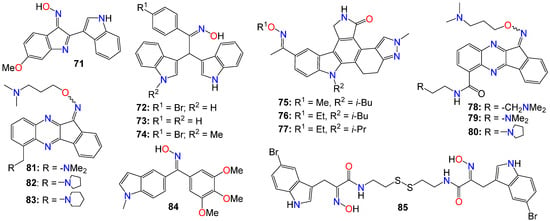
Figure 6. Illustrative examples of other antitumor indole derivatives bearing an oxime moiety.
A number of indoles psammaplin A derivatives were synthesized by replacing the o-bromophenol unit with an indole ring. Biochemical and cellular characterizations conducted on U937 and MCF-7 cells confirmed that many of these analogs exhibited more potent activities compared to the original natural product [124]. Furthermore, in addition to the well-documented dual HDAC and DNMT epigenetic inhibitory profile of the parent compound, some analogs, notably compound 85, also demonstrated inhibition of NAD+-dependent SIRT deacetylase enzymes. The structure–activity relationship (SAR) study provided insights into the mechanism of action underlying these multiple epigenetic ligands, and sets the stage for further structural exploration aimed at optimizing their pharmacological profiles [124]. In fact, enzymatic inhibition studies of HDAC1 and HDAC4 indicated that the C5-Br moiety could be substituted with other halogens (F, Cl, I) or oxygen-containing substituents (OMe, OBn), as these analogs displayed approximately equal potency. However, the absence of a substituent was found to be detrimental to the inhibitory activity [124]. Pharmacokinetic analyses revealed that compound 85 functions as a prodrug that rapidly transforms into a glutathione conjugate. The anticancer effects mediated by 85 seemed to involve the activation of distinct apoptosis pathways in cancer cells due to synergism between its inhibitory activities [124]. Notably, compound 85 demonstrated good tolerability in experimental mouse models, with a maximal tolerable dose higher than that of well-known HDAC inhibitors [124].
This entry is adapted from the peer-reviewed paper 10.3390/ijms242316854
This entry is offline, you can click here to edit this entry!