Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Glioblastoma (GBM) is the most aggressive and lethal primary brain tumor, bearing a survival estimate below 10% at five years, despite standard chemoradiation treatment. At recurrence, systemic treatment options are limited and the standard of care is not well defined, with inclusion in clinical trials being highly encouraged. So far, the use of immunotherapeutic strategies in GBM has not proved to significantly improve patients’ prognosis in the treatment of newly diagnosed GBM, nor in the recurrent setting.
- glioblastoma
- T cells
- tumor microenvironment
- immune check-point blockade
- vaccinations
- adoptive cell therapy
1. Targeting the PD-1/PD-L1 Axis, Alone or in Combination with Anti-CTLA4
The striking results of immunotherapy with checkpoint blockade in different cancer types have fueled numerous clinical studies in the field of neuro-oncology. After evidence in preclinical mouse models of TIL infiltration of brain tumors and truly promising data of tumor regression mediated by inhibition of specific co-stimulatory receptors (e.g., CTLA-4 and PD-1/PDL-1) [1][2][3], clinical trials testing checkpoint blockades in GBMs were designed. Different putative targets of immunotherapeutic strategies in GBM are depicted in Figure 1a. Table 1 summarizes the clinical trials with published results exploring the use of anti-PD-1/PD-L1 therapy in GBM in different phases of clinical development. The results of the early-phase part of the CheckMate 143 trial, which tested different ICB strategies in rGBM [4], led to the opening of the first randomized phase III study exploring the efficacy of anti-PD-1 agent nivolumab in rGBM [5]. A total of 439 patients with rGBM who progressed after the traditional Stupp regimen were randomized 1:1 to receive nivolumab or the anti-VEGF mAb bevacizumab, with OS as the primary end-point [5]. This study failed to demonstrate a statistically significant difference between treatment groups in terms of both median OS (mOS) and median progression-free survival (mPFS) (mOS: 9.8 and 10 months for nivolumab and bevacizumab, respectively; mPFS: 1.5 and 3.5 months for nivolumab and bevacizumab, respectively) [5]. Post hoc hypothesis-generating analyses conducted in previously pre-specified patient subgroups showed that patients with methylated MGMT promoter and without steroid use at baseline (n = 31) treated with nivolumab reached the longest mOS of 17 months [5]. The role of ICB in nGBM was further explored in the adjuvant setting by two different phase III trials: the CheckMate 498 trial randomized newly diagnosed GBM (nGBM) patients with unmethylated MGMT promoter to receive either nivolumab plus RT or standard TMZ plus RT, while the CheckMate 548 trial randomized methylated MGMT promoter patients to receive nivulomab plus TMZ and RT or placebo plus TMZ and RT [6][7]. Final results of CheckMate 498 showed an overlapping OS in the two groups (mOS 13.4 and 14.9 months for the experimental and control arm, respectively), with a similar incidence of grade 3/4 TRAEs (21.9% and 25.1% in the experimental and control arm, respectively) [6]. These findings may lead to a twofold speculative conclusion: on the one hand, knowing the poor efficacy of TMZ in unmethylated MGMT promoter patients [8], nivolumab may seem detrimental in this population; on the other hand, the researchers might conclude that TMZ continues to provide an albeit limited benefit in this population. Similarly, the CheckMate 548 trial did not meet its primary end-points, with no statistically significant differences in terms of both OS and PFS when nivolumab was added to standard TMZ and RT (mOS: 28.9 vs. 32.1 months and mPFS: 10.6 vs. 10.3 months in the experimental and placebo arms, respectively), even in the subgroup of patients without baseline corticosteroids [7]. Taking together the data that emerged from these phase III trials evaluating a global population of 1715 patients, of whom 496 were in the relapsed setting and the remaining at first diagnosis, it seems reasonable to conclude that the use of anti-PD-1 agents is not effective as a treatment for GBM in different phases of the disease course. However, as highlighted by phase 0 and translational trials, the timing of ICB may be crucial in influencing the quality, strength, and durability of the immune response. In the seminal paper by Cloughesy and colleagues, testing pembrolizumab in the neo\adjuvant setting in rGBM, patients randomized to receive immunotherapy in the neoadjuvant setting had a significantly prolonged survival compared to those in the adjuvant arm [9]. The significance of these outcomes were strengthened by translational data showing the upregulation of T cell and IFN-γ-related gene signatures and the enhanced clonal expansion of T cells (with focal induction of PD-L1) in the TME, underlying the potential capacity of ICB to stimulate/boost anti-tumor T cell responses and generate specific immunologic memory when given to an intact (not pre-treated) TME [9]. Despite the poor survival results coming from randomized clinical trials conducted so far, the chapter on immunotherapy with ICB in GBM may not yet be concluded. Several early-phase trials involving combinations of ICB, anti-angiogenic agents, or integration with other innovative immunotherapeutic strategies such as cellular or peptide vaccines are under intensive clinical investigation, with the aim of reverting immunoresistance, re-sensitizing GBM tumor cells, and finally avoiding immunotolerance (Table 2).
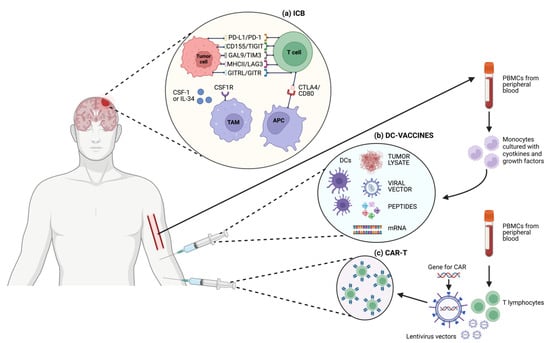
Figure 1. Schematic representation of different T cell-based immunotherapeutic strategies and targets in GBM. (a) ICB; (b) DC-vaccines; (c) CAR-T cells.
Table 1. Clinical trials (with published results) exploring the use of anti-PD-1/PD-L1 therapy in GBM.
| Title | Identifier | Study Design | Treatment | Population | Main Results | Main TRAEs |
|---|---|---|---|---|---|---|
| A Randomized Phase 3 Open Label Study of Nivolumab vs. Temozolomide in Combination with Radiation Therapy in Newly Diagnosed Adult Subjects with Unmethylated MGMT Glioblastoma (CheckMate 498) [6] | NCT02617589 | Phase 3 | A: nivolumab + radiotherapy B: temozolomide + radiotherapy |
Newly diagnosed methylated GBM | A: 13.0 months B: 14.2 months |
A: 21.9% G3-4 TRAEs B: 25.1% G3-4 TRAEs |
| Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter (CheckMate548) [7] | NCT02667587 | Phase 3 | A: nivolumab + temozolomide + radiotherapy B: placebo + temozolomide + radiotherapy |
Newly diagnosed methylated GBM | A: mPFS 10.6 months; mOS 31.3 months B: mPFS 10.3 months; mOS 33 months |
A: 52.4% G3-4 TRAEs B: 33.6% G3-4 TRAEs |
| Effect of Nivolumab vs. Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial [5] | NCT02017717 | Phase 3 | A: nivolumab B: bevacizumab |
Recurrent GBM | A: mOS: 9.8 months B: mOS 10 months |
A: 18.1% G3-4 TRAEs B: 15.2% G3-4 TRAEs |
| Circulating Immune Cells and Outcome Analysis from the Phase II Study of PD-L1 Blockade with Durvalumab for Newly Diagnosed and Recurrent Glioblastoma [10] | NCT02336165 | Phase 2 | A: RT + durvalumab B: durvalumab B2: durvalumab + bevacizumab B3: bevacizumab C: durvalumab + bevacizumab |
Newly diagnosed unmethylated GBM Recurrent GBM |
A: mOS 15.1 months B: mOS 6.7 months B2: mOS 8.7 months B3: mOS 9.3 months C: mOS 4.5 months |
Fatigue G>2: 18% Hyperlypase G>2: 8.8% Hyperamilase G>2: 5.6% Hypertension G>2: 5.6% |
| Tremelimumab and Durvalumab in Combination or Alone in Treating Patients with Recurrent Malignant Glioma | NCT02794883 | Phase 2 | A: tremelimumab B: durvalumab C: tremelimumab + durvalumab |
Recurrent GBM | A: mOS 7.2 months B: mOS 11.7 months C: mOS 7.7 months |
NR |
| Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma [11] | NCT02337491 | Phase 2 | A: pembrolizumab + bevacizumab B: pembrolizumab + placebo |
Recurrent GBM | A: mOS 8.8 months B: mOS 10.3 months |
A: 20% Hypertension G3 B: 10% Headache G3 |
| Nivolumab with DC Vaccines for Recurrent Brain Tumors | NCT02529072 | Phase 1 | A: pre-surgery nivolumab B: pre-surgery nivolumab + DC vaccine |
Grade III and Grade IV brain tumors | A: mOS 8 months B: mOS 15.3 months |
NR |
| Study of Cabiralizumab in Combination with Nivolumab in Patients With Selected Advanced Cancers | NCT02526017 | Phase 1 | cabiralizumab + nivolumab | GBM Other advanced solid tumors |
mOS 8 months | NR |
| Intracerebral administration of CTLA-4 and PD-1 immune checkpoint-blocking monoclonal antibodies in patients with recurrent glioblastoma: a phase I clinical trial [12] | NCT03233152 | Phase 1 | nivolumab (iv) + ipilimumab (intratumoral) | Recurrent GBM | mOS 37 weeks mPFS 11.7 weeks |
NR |
| Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial [13] | NCT04006119 | Phase 1 | Cemiplimab-rwlc + Ad-RTS-hIL-12 (intratumoral) + Veledimex (oral) | Recurrent GBM | mOS 8.9 months | NR |
GBM: glioblastoma; G: grade; TRAEs: treatment-related adverse events; mOS: median overall survival; mPFS: median progression-free survival; RT: radiotherapy; NR: not reported.
Table 2. Ongoing clinical trials exploring combinations of ICB, anti-angiogenic agents, or integration with other innovative immunotherapeutic strategies.
| Title | Identifier | Study Design | Treatment | Population | Recruitment Status |
|---|---|---|---|---|---|
| Neoadjuvant PD-1 Antibody Alone or Combined with Autologous Glioblastoma Stem-like Cell Antigen-Primed DC Vaccines (GSC-DCV) for Patients With Recurrent Glioblastoma: A Phase II, Randomized Controlled, Double Blind Clinical Trial | NCT04888611 | Phase 2 | A: camrelizumab + GSC-DCV B: camrelizumab + placebo |
Recurrent GBM | Recruiting |
| Testing the Use of the Immunotherapy Drugs Ipilimumab and Nivolumab Plus Radiation Therapy Compared to the Usual Treatment (Temozolomide and Radiation Therapy) for Newly Diagnosed MGMT Unmethylated Glioblastoma | NCT04396860 | Phase 2/3 | A: TMZ+RT B: TMZ+RT + ipilimumab + nivolumab |
Newly diagnosed unmethylated GBM Gliosarcoma |
Active, not recruiting |
| Efficacy and Safety Study of Neoadjuvant Efineptakin Alfa (NT-I7) and Pembrolizumab in Recurrent Glioblastoma [14] | NCT05465954 | Phase 2 | Efineptakin Alfa + pembrolizumab | Recurrent GBM | Recruiting |
| A phase II open-label, randomized study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol [15] | ISRCTN84434175 | Phase 2 | A: temozolomide + ipilimumab B: temozolomide |
Newly diagnosed GBM | Active, not recruiting |
| A Phase I/II Multicenter Trial Evaluating the Association of Hypofractionated Stereotactic Radiation Therapy and the Anti-Programmed Death-ligand 1 (PD-L1) Durvalumab (Medi4736) for Patients With Recurrent Glioblastoma (STERIMGLI) [16] | NCT02866747 | Phase 1/2 | A: hFSRT B: durvalumab + hFSRT |
Recurrent GBM | Active, not recruiting |
| Phase Ib/II Trial of Anti-PD-1 Immunotherapy and Stereotactic Radiation in Patients with Recurrent Glioblastoma | NCT04977375 | Phase 1/2 | pembrolizumab + stereotactic RT | Recurrent GBM | Recruiting |
| Pembrolizumab and Vorinostat Combined With Temozolomide for Newly Diagnosed Glioblastoma | NCT03426891 | Phase 1 | TMZ+RT + pembrolizumab + vorinostat | Newly diagnosed GBM | Active, not recruiting |
| A Phase II Study of Checkpoint Blockade Immunotherapy in Patients with Somatically Hypermutated Recurrent WHO Grade 4 Glioma | NCT04145115 | Phase 2 | nivolumab + ipilimumab | Recurrent Somatically Hypermutated GBM | Recruiting |
| Phase II Study of Pembrolizumab Plus SurVaxM for Glioblastoma at First Recurrence [17] | NCT04013672 | Phase 2 | A: SurVaxM + pembrolizumab (no prior anti-PD1 therapy) B: SurVaxM + pembrolizumab (prior anti-PD1 therapy) |
Recurrent GBM | Active, not recruiting |
| Phase II Study of Pembrolizumab (MK-3475) in Combination with Standard Therapy for Newly Diagnosed Glioblastoma | NCT03197506 | Phase 2 | A: neoadj and adj pembrolizumab (TMZ+RT) B: adj pembrolizumab (TMZ+RT) |
Newly diagnosed GBM | Recruiting |
| Pembrolizumab for Newly Diagnosed Glioblastoma (PERGOLA) | NCT03899857 | Phase 2 | TMZ+RT + pembrolizumab | Newly diagnosed GBM | Active, not recruiting |
| Avelumab in Patients With Newly Diagnosed Glioblastoma Multiforme (SEJ) [18] | NCT03047473 | Phase 2 | TMZ+RT + avelumab | Newly diagnosed GBM | Completed |
| Phase I/II Study to Evaluate the Safety and Clinical Efficacy of Atezolizumab (Anti-PD-L1) in Combination With Cabozantinib in Patients with Recurrent Glioblastoma (rGBM) | NCT05039281 | Phase 1/2 | cabozantinib + atezolizumab | Recurrent GBM | Recruiting |
| Combination of Checkpoint Inhibition and IDO1 Inhibition Together With Standard Radiotherapy or Chemoradiotherapy in Newly Diagnosed Glioblastoma. A Phase 1 Clinical and Translational Trial | NCT04047706 | Phase 1 | A (methylated): BMS-986205 + nivolumab (TMZ+RT) B (unmethylated): BMS-986205 + nivolumab (RT) |
Newly diagnosed GBM | Active, not recruiting |
| ASP8374 + Cemiplimab in Recurrent Glioma | NCT04826393 | Phase 1 | ASP8374 + cemiplimab | Recurrent GBM | Active, not recruiting |
| Pembrolizumab and a Vaccine (ATL-DC) for the Treatment of Surgically Accessible Recurrent Glioblastoma | NCT04201873 | Phase 1 | A: DC Tumor Cell Lysate Vaccine + pembrolizumab B: DC Tumor Cell Lysate Vaccine + placebo |
Recurrent GBM | Recruiting |
| Pharmacodynamic Study of Pembrolizumab in Patients With Recurrent Glioblastoma | NCT02337686 | Phase 2 | Pembrolizumab | Recurrent GBM | Active, not recruiting |
| An Open-Label, Multi-Center Trial of INO-5401 and INO-9012 Delivered by Electroporation (EP) in Combination With REGN2810 in Subjects With Newly-Diagnosed Glioblastoma (GBM) | NCT03491683 | Phase 1\2 | A (unmethylated): INO-5401 + INO-9012 + cemiplimab (TMZ+RT) B (methylated): INO-5401 + INO-9012 + cemiplimab + nivolumab (RT) |
Newly Diagnosed GBM | Active, not recruiting |
| A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells Combined with Checkpoint Inhibition for Patients With Resectable Recurrent Glioblastoma | NCT04003649 | Phase 1 | A: IL13Rα2-CAR-T + ipilimumab + nivolumab B: IL13Rα2-CAR-T + nivolumab C: IL13Rα2-CAR-T |
Recurrent GBM | Recruiting |
| Phase I Study of PD-L1 Inhibition With Avelumab and Laser Interstitial Thermal Therapy (LITT) in Patients with Recurrent Glioblastoma [19] | NCT03341806 | Phase 1 | A: avelumab B: avelumab + MRI-guided LITT |
Recurrent GBM | Completed |
GBM: glioblastoma; RT: radiotherapy; TMZ: temozolomide.
2. Other Co-Stimulatory and Co-Inhibitory Pathways
PD-1/TIGIT-axis emerged as putative immunotherapy targets in GBM in the analysis of TCGA datasets from 153 GBM patients. GBM patients with upregulated PD-1 and TIGIT genes and their cognate ligand genes displayed reduced OS and PFS compared to patients who did not upregulate the same genes [20]. In a GBM mouse model, treatment with anti-TIGIT/anti-PD-1 resulted in increased survival of the mice, reduced tumor volume, and increased immune infiltration in tumor lesions compared to single ICBs or isotype treatments [20]. A phase I clinical trial has been set up to study an anti-TIGIT antibody (AB154) in combination with anti-PD-1 therapy in rGBM (NCT04656535). In similar preclinical GBM models, dual blockade of TIM-3 and PD-1 or dual blockade of TIM-3 and BTLA [21] resulted in increased OS in treated mice compared to controls or single ICB treatment. Interestingly, both of these combination therapies proved more effective in increasing the proportion of cytokine-producing CD8+ T cells and in reducing the frequency of immunosuppressive Tregs within the tumor compared to single ICB treatment [21][22]. A phase I trial of anti-TIM3 in combination with anti-PD-1 treatment is now addressing the efficacy of this combination strategy in patients with rGBM (NCT03961971). Another marker of early T cell exhaustion, LAG3 (CD223), whose inhibition has been shown in preclinical models to be able to skew CD4+ cells away from the Treg phenotype [23], has been tested in a phase I trial for rGBM, alone or in combination with anti-PD-1 therapy (NCT02658981). Preliminary results of the first 44 patients enrolled indicated a mOS of 8 months for the anti-LAG3 monotherapy (with no dose-limiting toxicities (DLTs) for the highest safe dose of 800 mg) and 7 months for the combination therapy with anti-PD-1 [24]. Preclinical evidence demonstrated that another strategy for reshaping the TME, promoting effector T cell function, and inhibiting Tregs might be the activation of the glucocorticoid-induced TNFR-related gene (GITR) [25]. Nevertheless, GITR activating monotherapy has not proved effective in solid tumors [26], and combination strategies, together with another ICB (e.g., anti-PD-1) [27], chemotherapy [28], or RT [29], have been tested in pre-clinical and clinical trials to improve GITR agonism. An initial signal of the potential efficacy of these combination strategies came from a phase II trial for rGBM, where patients were treated with an anti-PD-1 plus a GITR agonist (INCAGN01876) together with fractionated stereotactic RT (FSRT) at disease recurrence (Cohort A) or, if amenable to surgery, patients were allocated to receive an anti-PD-1 plus INCAGN01876, with (Cohort B1) or without (Cohort B2) pre-surgical FSRT (NCT04225039). The combination of an anti-PD-1 plus INCAGN01876 and FSRT without surgical resection did not demonstrate efficacy in patients with rGBM, but interestingly, patients in Cohort B1 had significantly longer mPFS and mOS compared to patients in Cohort B2 (PFS 11.7 vs. 2.0 months, respectively; p = 0.0002, and mOS 20.1 vs. 9.4 months, respectively; p = 0.001), with a concomitant increase in inflammatory cytokine responses and proliferative T cells [30].
3. Vaccines
Vaccines are among the therapies that capitalize on the established efficacy of the immune system in fighting cancer; they work by activating the adaptive immune system, particularly T cells, against antigens that might be set up and injected from outside to be carried on the membrane of APCs, or activated APCs can be administered directly [31]. The optimal source of antigens to be used for APC loading is still controversial, as the “perfect” antigen should bear tumor specificity in order to avoid autoimmune attacks on healthy cells but also be broadly present at the tumor site, allowing targeting of as many cancer cell clones as possible. In view of this, the researchers can distinguish so-called tumor-associated antigens (TAAs), ubiquitous non-mutated proteins that are upregulated in tumors compared to normal tissues (e.g., EGFR, IL-13Rα2, and gp100), and tumor-specific antigens (TSAs), produced and expressed only by tumor cells (e.g., EGFRvIII). For example, taking into account GBM therapy, a phase III trial evaluating Rindopepimut, a peptide vaccine targeting the EGFR mutation EGFRvIII, did not show any survival benefit in patients with nGBM [32]. In contrast, favorable results were reported for a peptide vaccine against the surviving molecule, a TAA, namely SurVaxM, tested in a phase II trial in resected nGBM, showing a mPFS of 11.4 months and a mOS of 25.9 months [33].
Here, the researchers will focus on so-called DC-vaccines, specifically describing the results of the most recent phase II and III randomized clinical studies. As previously shown, DCs might be distinguished into conventional/myeloid DC 1-2 (cDC1, cDC2) and plasmacytoid DC (pDC); the cDC1 subtype appears to have a higher affinity for activating CD8+ T cells, while cDC2 is for CD4+ T cells [34]. In manufacturing DC-based vaccines, it is unknown which subsets or combinations are most efficient; nevertheless, the most common technology involves the production of monocyte-derived DC ex vivo by in vitro stimulation of monocytes isolated from peripheral blood mononuclear cells (PBMCs). The specific methodology to isolate and culture/differentiate monocytes into cDCs is still controversial, with different cell culture media, serum sources, and required concentrations of GM-CSF and IL-4 used in different studies [35]. Also, the sources of antigens used to activate autologous ex vivo-generated DC may be different in nature, such as whole-tumor lysate, antigenic peptides, viral transfection, or messenger RNA (mRNA) electroporation. A schematic representation of the DC vaccine strategies is depicted in Figure 1b. The development of trials with DC vaccines in GBM has been hampered by issues common to other solid tumors, such as the selection of the best antigen to stimulate an effective and sustained immune response, the timing of administration (e.g., neo-adjuvant setting or advanced disease), the expansive cost of manufacturing, and the long turnaround time. While specific problems in developing DC vaccines for GBM include the low sample size given the disease’s low incidence, severely limiting the ability to conduct large trials with control arms, and the short disease median OS, especially for relapsed GBM, greatly reducing the time window available to produce and demonstrate the efficacy of therapies aiming to activate the immune system [36]. A list of trials testing DC vaccination in GBM with reported results is displayed in Table 3. The phase III trial assessing tumor lysate-loaded DC vaccine (DCVax-L) in patients with nGBM, although formally meeting its primary endpoint, with a mOS of 19.3 versus 16.5 months (HR = 0.80; 98% CI, 0.00–0.94; p = 0.002) for patients receiving DCVax-L and placebo plus TMZ, respectively, was very controversially conducted [37]. Important limitations, both in the nature and methodology of this study, need to be highlighted. Firstly, in the original trial design, the primary end-point was PFS determined by MRI, but due to difficulties in distinguishing progressive disease from pseudo-progression related to immune-related necrosis or inflammation, OS was used for the definitive statistical analyses as the primary end-point. Moreover, this study was presented as randomized, double-blind, with a crossover design, but due to the high number of patients accessing the crossover phase, the placebo group was depleted, and the survival analyses were conducted using a post hoc external control population for whom individual patient level data were not available, and thus a propensity score analysis was not feasible. Thus, taking all this into account, the trial should not be considered to answer the question on DCVax-L activity in GBM, and its post hoc conclusive analyses are to be accounted for only as hypothesis-generating [38].
Table 3. Clinical trials (with published results) testing DC vaccination in GBM.
| Title | Identifier | Study Design | Vaccine Production | Population | Main Results | Treatment Related Adverse Events (TRAEs) |
|---|---|---|---|---|---|---|
| A Phase III Clinical Trial Evaluating DCVax®-L, Autologous Dendritic Cells Pulsed With Tumor Lysate Antigen For The Treatment Of Glioblastoma Multiforme (GBM) [37] | NCT00045968 | Phase 3 | Tumor lysate | Newly diagnosed GBM Recurrent GBM |
nGBM mOS: 19.3 months rGBM mOS: 13.2 months |
G 3\4 TRAEs: 2 cases of intracranial edema (G3), 1 case of nausea (G3), and 1 case of lymph node infection (G3) |
| Phase II Trial of Autologous Dendritic Cells Loaded With Autologous Tumor Associated Antigens (AV-GBM-1) as an Adjunctive Therapy Following Primary Surgery Plus Concurrent Chemoradiation in Patients With Newly Diagnosed Glioblastoma [39] | NCT03400917 | Phase 2 | Lysate of irradiated autologous tumor-initiating cells | Newly diagnosed GBM | mOS: 16 months 2-years OS: 27% mPFS: 10.4 months |
G 3\4 TRAEs: none |
| A Randomized, Double-blind, Controlled Phase IIb Study of the Safety and Efficacy of ICT-107 in Newly Diagnosed Patients With Glioblastoma Multiforme (GBM) Following Resection and Chemoradiation [40] | NCT01280552 | Phase 2 | Synthetic peptide epitopes targeting GBM associated antigens MAGE-1, HER-2, AIM-2, TRP-2, gp100, and IL13Rα2 |
Newly diagnosed GBM | mOS: 17 months (vs. 15 months, NS) mPFS: 11.2 months (vs. 9 months, p = 0.011) |
G 3\4 TRAEs: no difference from the control group (G3 nervous system disorders, fatigue, WBC decrease, and infections) |
| A Phase II Feasibility Study of Adjuvant Intra-Nodal Autologous Dendritic Cell Vaccination for Newly Diagnosed Glioblastoma Multiforme | NCT00323115 | Phase 2 | Tumor lysate | Newly diagnosed GBM | mOS: 28 (15-44) months mPFS: 9.5 (5-41) months |
N.A. |
| Phase I Trial of Vaccination With Autologous Dendritic Cells Pulsed With Lysate Derived From an Allogeneic Glioblastoma Stem-like Cell Line for Patients With Newly Diagnosed or Recurrent Glioblastoma [41] | NCT02010606 | Phase 1 | Lysate from an allogeneic stem-like cell line | Newly diagnosed GBM Recurrent GBM |
nGBM mOS: 20.36 months mPFS: 8.75 months rGBM mOS: 11.97 months mPFS: 3.23 months |
G 3\4 TRAEs: none |
| A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma [42] | N.A. | Phase 1 | Tumor lysate | Newly diagnosed GBM Recurrent GBM |
nGBM mOS: 27.7 months mPFS: 4.8 months rGBM mOS: 10.9 months mPFS: 1.9 months |
G 3\4 TRAEs: none |
| Anti-Tumor Immunotherapy Targeted Against Cytomegalovirus in Patients With Newly-Diagnosed Glioblastoma Multiforme During Recovery From Therapeutic Temozolomide-induced Lymphopenia | NCT00639639 | Phase 1 | CMV pp65 | Newly diagnosed GBM | ATTAC-GM mOS: 37.7 months ATTAC-Td mOS: 38.3 months |
N.A. |
| Evaluation of Overcoming Limited Migration and Enhancing Cytomegalovirus-specific Dendritic Cell Vaccines With Adjuvant TEtanus Pre-conditioning in Patients With Newly-diagnosed Glioblastoma | NCT02366728 | Phase 2 | CMV pp65 | Newly diagnosed GBM | Arm1 (unpulsed DC): mOS 16 months Arm2 (Td pre-conditioning): mOS 20 months Arm3 (Basiliximab + Td pre-conditioning): mOS 19 months |
SAEs: Arm 1: 1\27 pts (lung infection) Arm 2: 4\28 pts (urinary infection, nervous system, and psychiatric disorders) Arm 3: 2\9 pts (colon perforation, nervous system disorders) |
nGBM: newly diagnosed glioblastoma; rGBM: recurrent glioblastoma; G: grade; TRAEs: treatment-related adverse events; mOS: median overall survival; mPFS: median progression-free survival; N.A.: not applicable; SAEs: severe adverse events.
As GBM cells might contain sequences and express the viral gene products of CMV [43], investigators at Duke University designed a set of clinical trials assessing a vaccination with CMV pp65-specific autologous DCs. The ATTAC trial (testing the pre-conditioning of the vaccine site with tetanus/diphtheria (Td) toxoid) [NCT00639639], the ATTAC-GM (expanded cohort receiving GM-CSF containing autologous CMV DC vaccines with dose-intensified TMZ) [NCT00639639], and the ELEVATE trial [NCT02366728] [44]. The vaccination was administered with TZM in nGBM after surgical resection and chemoradiation and proved very encouraging 5-year OS rates of 33.3% (mOS 38.3 months; 95% CI, 17.5–∞) and 36.4% (mOS 37.7 months; 95% CI, 18.2–109.1) for ATTAC-Td and ATTAC-GM, respectively, without significant TRAEs [44]. In the confirmatory double-blind randomized phase II trial (ELEVATE), the 3-year OS for the Td arm was 34% (95% CI, 19–63%) compared to 6% (95% CI, 1–42%) in the unpulsed DCs arm [45]. Translational analyses in the ATTAC study showed a steady increase in the Treg population after the first dose of maintenance TMZ, followed by an increase in peripheral CD8+ T cells and the CD8+:Treg ratio after pp65 vaccination [46]. Therefore, in the ELEVATE trial, the investigators added a second experimental arm using the anti-Treg monoclonal antibody Basiliximab (anti-IL-2rα) in order to test whether inhibition of the initial increase in the Treg pool may serve as a boost for the T-mediated response induced by the pp65 DC vaccine. Preliminary data did not show a survival advantage in the Basiliximab arm compared with patients treated with DC vaccine plus Td pre-conditioning, with a mOS of 19 (16.7–25.6) and 20 (10.26–NA) months, respectively [NCT02366728].
While promising, therapies in GBM based on DC vaccines are far from clinical practice, and the researchers still await the results of larger, more advanced-phase trials to demonstrate a substantial clinical benefit. Translational analyses will be crucial to understanding how to develop a vaccine able to induce strong and long-lasting T-cell-mediated immune activation against GBM cells. Many early-phase clinical trials are attempting to increase the immunogenic potential of the DC vaccines in GBM; specifically, the most promising ongoing strategies are testing the combination of DC vaccines with ICB or local therapies such as RT or medicated wafers [42][47].
4. Adoptive Cell Therapy and Chimeric Antigen Receptor T Cell Therapy
Adoptive cell therapy (ACT), also known as cellular immunotherapy, is a form of treatment that uses immune cells to target and potentially eliminate cancer cells. ACT approaches range from isolating a patient’s immune cells (e.g., T lymphocytes, DCs, or NK cells), expanding their number in vitro, and then reinfusing them, to genetically engineered (via gene therapy) autologous immune cells such as chimeric antigen receptor T (CAR-T) cells. In the former case, tumor-infiltrating immune cells are obtained from the enzymatically digested tumor specimens and activated ex vivo; then specific clones, presenting with high avidity for tumor antigens, are selected and expanded before reinfusion. In the latter, patients’ T cells are equipped with engineered synthetic receptors, known as CARs, to recognize and eliminate tumor cells expressing a specific target antigen; importantly, this binding is independent from the MHC receptor, thus possibly overcoming the limitation of the previously described form of ACT. Here, the researchers will focus on treatment strategies based on CAR-T cells, the most investigated and clinically meaningful ACT application of autologous T cells; a schematic representation of the generation of CAR-T cells is depicted in Figure 1c. CARs are composed of four main components: an extracellular target antigen-binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains. Indeed, different subsequent generations of CARs, based on the quality and quantity of co-stimulatory intracellular receptors with tyrosin-kinase activity, in series with the CD3ζ have been designed and proposed for clinical testing. After the success of CAR-T cell-based approaches in hematological malignancies and the approval for clinical use in several countries of CAR-T therapy for the treatment of relapsed or refractory lymphomas, leukemia, or multiple myeloma [48][49][50][51], this specific ACT application has been under investigation in many different solid tumors. Nevertheless, specific issues characterizing solid tumors in contrast to hematological malignancies, such as inefficient trafficking and infiltration at the tumor site, immunosuppressive TME, and antigen heterogeneity, have been limiting the clinical applicability of this treatment strategy [52][53]. Overexpression of mutant EGFR variant III (EGFRvIII), present in about 30% of nGBM [54], has been recognized as a tumor-specific, oncogenic epitope able to mediate increased cellular growth, invasion and angiogenesis, and resistance to chemoradiation in GBM cells [55]. Thus, EGFRvIII has been selected as a putative target and is actually the most explored in clinical trials for CAR-T cells against GBM cells. Nevertheless, taking into account the three completed phase I clinical trials testing EGFRvIII CAR-T cells in GBM (NCT02209376, NCT02664363, and NCT01454596), no clear radiological responses were observed. It is worth noting that in the study by O’Rourke and colleagues, 1 patient had residual stable disease for over 18 months after a single dose of CAR-T EGFRvIII cells; translational analyses on tissue specimens after surgery (available for 7 of the enrolled patients) showed antigen decrease at the site of active GBM in 5 out of 7 patients and increased expression of inhibitory molecules and Tregs, highlighting how important it would be for future studies to understand how to overcome immune changes in the TME [56]. Another tumor-specific antigen, Interleukin 13 receptor α2 (IL13Rα2), linked to GBM poor prognosis, has been used to engineer IL13Ra2-specific CAR-T cells to be infused via a catheter/reservoir system into the resection cavity in a pilot safety and feasibility trial that demonstrated fair tolerability and transient anti-glioma responses in 2 out of 3 patients treated [57]. Thus, a phase I trial has been designed with modified co-stimulation and the linker of the IL13Ra2-specific CAR-T to improve antitumor efficacy [58]. Multiple infusions of CAR-T cells were administered both directly into the tumor cavity and into the ventricular system; no grade 3 or higher TRAEs were observed, and a stable regression of tumor localizations (both intracranial and spinal) lasted for 7.5 months after the first CAR-T infusion [58]. Given that overexpression of human epidermal growth factor receptor 2 (HER2) is well known to drive carcinogenesis in several different solid tumors (such as breast cancer, gastric cancer, CRC, and NSCLC) and that it is also found in around 80% of GBMs [59], HER2 CAR-T cell therapy has been extensively explored, with mixed clinical results and concerns about toxicities due to off-target effects [60]. In a phase I trial of HER2-positive GBM testing HER2-CAR CMV pp65-bispecific cytotoxic T lymphocytes, no DLTs were observed, 1 patient experienced PR for more than 9 months, and 7 patients had SD lasting 8 weeks to 29 months, with a mOS of 11.1 months (95% CI, 4.1–27.2 months), leaving an open window for further exploring this therapeutic target [61].
This entry is adapted from the peer-reviewed paper 10.3390/cancers16030603
References
- Wainwright, D.A.; Chang, A.L.; Dey, M.; Balyasnikova, I.V.; Kim, C.K.; Tobias, A.; Cheng, Y.; Kim, J.W.; Qiao, J.; Zhang, L.; et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 2014, 20, 5290–5301.
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167.
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135.
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686.
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010.
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; Van Den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023, 25, 123–134.
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022, 24, 1935–1949.
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; De Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003.
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486.
- Nayak, L.; Standifer, N.; Dietrich, J.; Clarke, J.L.; Dunn, G.P.; Lim, M.; Cloughesy, T.; Gan, H.K.; Flagg, E.; George, E.; et al. Circulating Immune Cell and Outcome Analysis from the Phase II Study of PD-L1 Blockade with Durvalumab for Newly Diagnosed and Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 2567–2578.
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; De Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057.
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296.
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.A.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro Oncol. 2022, 24, 951–963.
- Webb, M.; Sener, U.; Burns, T.; Twohy, E.; Kizilbash, S.H.; Ruff, M.; Uhm, J.; Galanis, E.; D’Andre, S.; Riviere-Cazaux, C.; et al. Efficacy and safety study of neoadjuvant efineptakin alfa (NT-I7) and pembrolizumab in recurrent glioblastoma. J. Clin. Oncol. 2023, 41 (Suppl. S16).
- Brown, N.F.; Ng, S.M.; Brooks, C.; Coutts, T.; Holmes, J.; Roberts, C.; Elhussein, L.; Hoskin, P.; Maughan, T.; Blagden, S.; et al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: The Ipi-Glio trial protocol. BMC Cancer 2020, 20, 198.
- Pouessel, D.; Ken, S.; Gouaze-Andersson, V.; Piram, L.; Mervoyer, A.; Larrieu-Ciron, D.; Cabarrou, B.; Lusque, A.; Robert, M.; Frenel, J.-S.; et al. Hypofractionated Stereotactic Re-irradiation and Anti-PDL1 Durvalumab Combination in Recurrent Glioblastoma: STERIMGLI Phase I Results. Oncologist 2023, 28, e817–e825.
- Ahluwalia, M.; Peereboom, D.; Rauf, Y.; Schilero, C.; Ciolfi, M.; Ciesielski, M.; Fenstermaker, R. Phase II study of pembrolizumab plus SurVaxM for glioblastoma at first recurrence. J. Clin. Oncol. 2020, 38 (Suppl. S15), TPS2581.
- Ahluwalia, M.; Peereboom, D.; Rauf, Y.; Schilero, C.; Ciolfi, M.; Ciesielski, M.; Fenstermaker, R. Avelumab in newly diagnosed glioblastoma. Neurooncol. Adv. 2021, 3, vdab118.
- Hormigo, A.; Chiu, D.; Hahn, M.; Qi, J.; Lee, B.; Mandeli, J.; Ghatan, S.; Hadjipanayis, C.; Yong, R.; Germano, I.; et al. CTIM-09. Phase I study of PD-L1 inhibition with avelumab and laser interstitial thermal therapy in patients with recurrent glioblastoma. Neuro-Oncology 2021, 23 (Suppl. S6), vi51.
- Raphael, I.; Kumar, R.; McCarl, L.H.; Shoger, K.; Wang, L.; Sandlesh, P.; Sneiderman, C.T.; Allen, J.; Zhai, S.; Campagna, M.L.; et al. TIGIT and PD-1 Immune Checkpoint Pathways Are Associated With Patient Outcome and Anti-Tumor Immunity in Glioblastoma. Front. Immunol. 2021, 12, 637146.
- Choi, J.; Medikonda, R.; Saleh, L.; Kim, T.; Pant, A.; Srivastava, S.; Kim, Y.-H.; Jackson, C.; Tong, L.; Routkevitch, D.; et al. Combination checkpoint therapy with anti-PD-1 and anti-BTLA results in a synergistic therapeutic effect against murine glioblastoma. Oncoimmunology 2021, 10, 1956142.
- Kim, S.T.; Klempner, S.J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, K.-M.; Lee, J. Correlating programmed death ligand 1 (PD-L1) expression, mismatch repair deficiency, and outcomes across tumor types: Implications for immunotherapy. Oncotarget 2017, 8, 77415–77423.
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080.
- Lim, M.; Ye, X.; Nabors, L.B.; Piotrowski, A.; Ahluwalia, M.; Desai, A.; Walbert, T.; Fisher, J.; Desideri, S.; Sims, M.; et al. Updated phase I trial of anti-LAG-3 or anti-CD137 alone and in combination with anti-PD-1 in patients with recurrent GBM. J. Clin. Oncol. 2019, 37 (Suppl. S15), 2017.
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10.
- Davar, D.; Zappasodi, R. Targeting GITR in cancer immunotherapy—There is no perfect knowledge. Oncotarget 2023, 14, 614–621.
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235.
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 2014, 12, 36.
- Schoenhals, J.E.; Cushman, T.R.; Barsoumian, H.B.; Li, A.; Cadena, A.P.; Niknam, S.; Younes, A.I.; Caetano, M.D.S.; Cortez, M.A.; Welsh, J.W. Anti-glucocorticoid-induced Tumor Necrosis Factor-Related Protein (GITR) Therapy Overcomes Radiation-Induced Treg Immunosuppression and Drives Abscopal Effects. Front. Immunol. 2018, 9, 2170.
- Pouessel, D.; Ken, S.; Gouaze-Andersson, V.; Piram, L.; Mervoyer, A.; Larrieu-Ciron, D.; Cabarrou, B.; Lusque, A.; Robert, M.; Frenel, J.-S.; et al. PD1 inhibition and GITR agonism in combination with fractionated stereotactic radiotherapy for the treatment of recurrent glioblastoma: A phase 2, multi-arm study. J. Clin. Oncol. 2023, 41 (Suppl. S16), 2004.
- Saxena, M.; Van Der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378.
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385.
- Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Figel, S.A.; Mechtler, L.L.; Peereboom, D.M.; Hutson, A.D.; Withers, H.G.; et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2023, 41, 1453–1465.
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408.
- Chometon, T.Q.; Siqueira, M.D.S.; Sant´anna, J.C.; Almeida, M.R.; Gandini, M.; Martins De Almeida Nogueira, A.C.; Antas, P.R.Z. A protocol for rapid monocyte isolation and generation of singular human monocyte-derived dendritic cells. PLoS ONE 2020, 15, e0231132.
- Hotchkiss, K.M.; Batich, K.A.; Mohan, A.; Rahman, R.; Piantadosi, S.; Khasraw, M. Dendritic cell vaccine trials in gliomas: Untangling the lines. Neuro Oncol. 2023, 25, 1752–1762.
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination With Extension of Survival Among Patients With Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121.
- Preusser, M.; van den Bent, M.J. Autologous tumor lysate-loaded dendritic cell vaccination (DCVax-L) in glioblastoma: Breakthrough or fata morgana? Neuro Oncol. 2023, 25, 631–634.
- Bota, D.A.; Taylor, T.H.; Piccioni, D.E.; Duma, C.M.; LaRocca, R.V.; Kesari, S.; Carrillo, J.A.; Abedi, M.; Aiken, R.D.; Hsu, F.P.K.; et al. Phase 2 study of AV-GBM-1 (a tumor-initiating cell targeted dendritic cell vaccine) in newly diagnosed Glioblastoma patients: Safety and efficacy assessment. J. Exp. Clin. Cancer Res. 2022, 41, 344.
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807.
- Hu, J.L.; Omofoye, O.A.; Rudnick, J.D.; Kim, S.; Tighiouart, M.; Phuphanich, S.; Wang, H.; Mazer, M.; Ganaway, T.; Chu, R.M.; et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent Glioblastoma. Clin. Cancer Res. 2022, 28, 689–696.
- Rudnick, J.D.; Sarmiento, J.M.; Uy, B.; Nuno, M.; Wheeler, C.J.; Mazer, M.J.; Wang, H.; Hu, J.L.; Chu, R.M.; Phuphanich, S.; et al. A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma. J. Clin. Neurosci. 2020, 74, 187–193.
- Rahman, M.; Dastmalchi, F.; Karachi, A.; Mitchell, D. The role of CMV in glioblastoma and implications for immunotherapeutic strategies. Oncoimmunology 2019, 8, e1514921.
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303.
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Reproducibility of outcomes in sequential trials using CMV-targeted dendritic cell vaccination for glioblastoma. J. Clin. Oncol. 2022, 40 (Suppl. S16), 2005.
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E.; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909.
- Miller, A.; Kosaloglu-Yalcin, Z.; Westernberg, L.; Montero, L.; Bahmanof, M.; Frentzen, A.; Logandha Ramamoorthy Premlal, A.; Greenbaum, J.; Seumois, G.; Habbaba, R.; et al. A phase 1b study of personalized neoantigen vaccine plus pembrolizumab in adults with advanced cancer. J. Clin. Oncol. 2021, 39 (Suppl. S15), 2615.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544.
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56.
- Shah, N.N.; Lee, D.W.; Yates, B.; Yuan, C.M.; Shalabi, H.; Martin, S.; Wolters, P.L.; Steinberg, S.M.; Baker, E.H.; Delbrook, C.P.; et al. Long-Term Follow-Up of CD19-CAR T-Cell Therapy in Children and Young Adults With B-ALL. J. Clin. Oncol. 2021, 39, 1650–1659.
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716.
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929.
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159.
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma. Front. Oncol. 2015, 5, 5.
- Learn, C.A.; Hartzell, T.L.; Wikstrand, C.J.; Archer, G.E.; Rich, J.N.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin. Cancer Res. 2004, 10, 3216–3224.
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984.
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072.
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569.
- Mineo, J.-F.; Bordron, A.; Baroncini, M.; Maurage, C.-A.; Ramirez, C.; Siminski, R.-M.; Berthou, C.; Dam Hieu, P. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J. Neurooncol. 2007, 85, 281–287.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851.
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101.
This entry is offline, you can click here to edit this entry!