1. Introduction
SIRTs, which comprise the Silent Information Regulator 2 family of ancient proteins, are ubiquitous throughout all domains of life [
1]. Seven SIRTs characterize mammals, numbered SIRT1 through SIRT7. SIRTs are distinctive post-translational modification enzymes that use NAD+ as a co-substrate to eliminate acyl groups from lysine residues. SIRTs display an effect on a wide range of substrates and significant metabolic processes. SIRTs target a broad spectrum of cellular proteins for post-translational modification via acetylation (e.g., SIRT1, 2, 3, and 5) or ADP-ribosylation (SIRT4 and 6). SIRT7 regulates the transcription of RNA polymerase I [
2]. A diverse range of lipid acyl groups (e.g., acetyl, glutaryl, malonyl, succinyl, or long-chain acyl groups) are transferred from substrate proteins to the ADP-ribose portion of NAD+ via the distinctive NAD+-dependent protein deacylase activity of SIRTs [
1,
3,
4,
5,
6]. Due to the critical significance of NAD+ in the cellular metabolism, SIRTs are able to regulate the function of a wide variety of protein substrates (including histones, transcription factors, metabolic enzymes, and cell membrane proteins) exclusively relying on NAD+.
SIRT1, the most conserved SIRT in mammals, is predominantly localized in the nucleus but can transition between the cytosol and nucleoplasm in response to various environmental stimuli [
7,
8]. SIRT6 and SIRT7 are also nuclear proteins. SIRT7 is significantly enriched in the nucleolus, whereas SIRT6 is a chromatin-bound protein which is localized within the nucleus [
9,
10]. SIRT3, SIRT4, and SIRT5, three additional mammalian SIRTs, are located in the mitochondrial matrix, where they are involved in a variety of survival and metabolic processes associated with mitochondrial activity [
11,
12]. A considerable segment of SIRT5 is located outside of mitochondria, in the cytosol. It regulates glycolysis by demalonylating essential glycolytic enzymes. SIRT2, a protein which can also migrate between the cytosol and the nucleus, is predominantly cytosolic [
11,
12]. Existing research has focused extensively on the significance of SIRTs in relation to aging, age-related conditions and diseases, and nutrient sensing [
13,
14]. However, recent findings indicate that this family of distinctive enzymes also plays a crucial role in regulating stem cell biology [
1]. Different SIRTs have different effects on SCs depending on the type of cell or stage in response to different environmental stimuli, even though they all depend on cellular NAD+. Embryogenesis and pluripotent SCs can be regulated by nuclear and cytosolic SIRTs through various mechanisms, including epigenetics, redox homeostasis, metabolism regulation, cellular stress response, and the control of pluripotency factors. SIRTs safeguard against stress-induced and aging-induced depletion of adult stem cells by preserving their capacity for self-renewal, quiescence, and regeneration [
1].
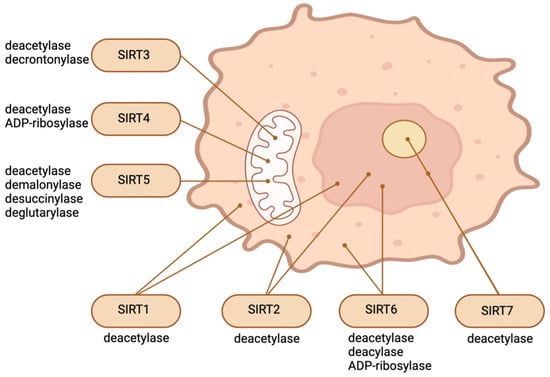
Figure 1 illustrates the main enzymatic functions and intracellular location of SIRTs.
Figure 1. Intracellular location and main enzymatic activities of SIRTs. The figure was partly created with BioRender.com.
In light of the significance and far-reaching consequences associated with SIRTs’ activity, it is critical to comprehend the factors that govern their expression and activity. SIRT1, the most thoroughly investigated constituent of the SIRT family, has demonstrated regulation across various levels. While the precise mechanisms governing other SIRTs remain poorly comprehended, it seems that they operate within a regulatory environment that is similarly complex [
15]. SIRT regulatory factors can be classified into four main categories [
15,
16,
17]: (1) Regulating the transcription of multiple SIRTs can be achieved by modulating their nucleocytoplasmic transport. CCAAT-enhancer-binding protein-α and E2F transcription factor 1 promote the transcription of SIRT1, whereas p53 and Hypermethylated in cancer 1 inhibit it [
15]. (2) The levels of available NAD substrate have an impact on the activity of SIRTs. AMP-activated protein kinase increases the expression of nicotinamide phosphoribosyl transferase to control NAD levels [
15]. (3) SIRT activity can be changed at the protein level. SIRT1, for instance, is activated via a protein–protein interaction when the active regulator of SIRT1 binds directly to it, and sumoylation occurs at lysine 734. In contrast, direct binding with Deleted in Breast Cancer 1 (DBC1) inhibits SIRT1. Additionally, the post-translational modification mechanisms of SIRTs are diverse. The N- and C-terminal regions of the various SIRTs, which extend beyond the catalytic domains, mediate this regulation. These extensions are subject to various post-translational modifications, such as methylation, sumoylation, phosphorylation, and proteolytic cleavage [
15,
16]. (4) Additionally, the half-life of SIRT mRNAs is regulated. Binding the human antigen R RNA-binding protein to SIRT1 mRNA, for instance, prolongs the half-life of the latter. On the contrary, the implementation of miR-34a, miR-132, or miR-217 into SIRT1 leads to the translational repression of translation [
15,
16]. In addition, natural food products contain SIRT-activating compounds, which are small molecules responsible for regulating the expression of SIRTs. These are catechins, resveratrol, quercetin, berberine, fisetin, and curcumin [
17].
2. The Role of the SIRTs in the Control of Stem Cells
SIRT1. In comparison to adult tissues and cells, SIRT1 is significantly overexpressed in preimplantation embryos and ESCs [
18]. It is essential for the maintenance of healthy development and embryogenesis [
19,
20,
21]. SIRT1 is primarily responsible for silencing genes via heterochromatin formation. Supporting the creation of facultative heterochromatin, SIRT1 deacetylates H4K6, H3K9, and H3K56 preferentially. By acetylating and recruiting histone H1 to chromatin, SIRT1 is capable of enhancing local compactness. It is also capable of deacetylating nonhistone targets that participate in the formation of heterochromatin. SIRT1 is involved in the maintenance of cellular homeostasis during stress [
22].
Accumulating evidence suggests that the deacetylation activity of SIRT1 regulates embryogenesis, development, and the maintenance of ESC pluripotency through hierarchical mechanisms [
1]. SIRT1 in stem cells deacetylates histones, the epigenetic regulator DNMT3L, a critical component of the core pluripotency network OCT4, and a tumor suppressor p53 [
23,
24,
25]. SIRT1 regulates the differentiation capacity of mouse ESCs and the expression of imprinted and germline genes [
23]. Through the direct deacetylation of histones, SIRT1 inhibits the expression of differentiation-associated genes in ESCs. Reducing SIRT1 during embryonic development leads to the reactivation of those developmental genes [
26]. Additionally, SIRT1 inhibits RAR-mediated activation of differentiation genes in mouse ESCs [
20]. SIRT1 is essential for the maintenance of robust pluripotent ESCs as well. SIRT1 facilitates the translocation of p53 from the nucleus to the cytoplasm as a consequence of endogenous ROS damage. SIRT1 exposes ESCs to apoptosis induced by mitochondrial p53 in mice, while also impeding the suppression of Nanog expression mediated by nuclear p53 [
27]. In sum, SIRT1 functions as an important regulator in coordinating metabolic and epigenetic signaling pathways to ensure the maintenance of pluripotent ESCs and normal embryogenesis through the deacetylation of key regulators.
Stem cells, such as mouse ESCs and iPSCs, exhibit high expression levels of both SIRT1 and c-Myc. SIRT1 increases the stability of c-Myc in these stem cells by deacetylating it; this is likely the result of a reported exchange of K63-linked versus K48-linked polyubiquitination chains [
18,
28]. Enhanced c-Myc stability promotes the transcription of c-Myc target genes in stem cells, like Usp22, Mat2a, Smpdl3b, and Tert. Elevated MAT2A expression in mouse ESCs stimulates the synthesis of SAM from methionine; this, in turn, elevates the H3K4me3 content of pluripotent genes, thereby stimulating their expression [
18,
19]. This process is critical for the maintenance of pluripotent stem cells. Additionally, c-Myc raises the production of SMPDL3B in mouse ESCs. This changes the sphingolipids on the plasma membrane and affects signaling pathways that promote neuronal differentiation as well as the fluidity of the membrane. C-Myc increases the transcription of Tert in post-reprogrammed iPSCs in order to facilitate telomere elongation [
29]. In the absence of SIRT1, iPSCs amass chromosomal aberrations and exhibit telomeric heterochromatin de-repression. Consequently, SIRT1 exerts a positive regulatory effect on TERT expression through the enhancement of c-Myc protein stability (
Figure 2) [
29].
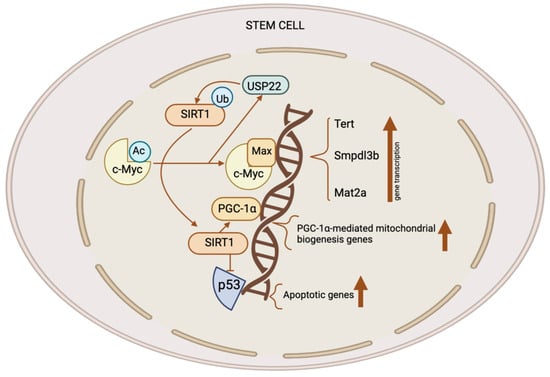
Figure 2. The role of the SIRT1–c-Myc axis in the control of stem cells. Both SIRT1 and c-Myc have significant expression levels in several types of stem cells. SIRT1 removes acetyl groups from c-Myc, leading to enhanced stability. When c-Myc is more stable, it increases the transcription of c-Myc target genes like Usp22, Mat2a, Tert, and Smpdl3b. The upregulation of MAT2A leads to the synthesis of SAM, resulting in an elevation of H3K4me3 levels on pluripotent genes and subsequently promoting their expression. This activity is crucial for the preservation of pluripotent stem cells. C-Myc also stimulates the production of SMPDL3B to modify sphingolipids on the plasma membrane, affecting the flexibility of the membrane and the signaling pathways which are involved in the process of neuronal development. C-Myc stimulates the process of transcribing Tert, which, in turn, enhances the elongation of telomeres. C-Myc enhances the excessive production of USP22, an enzyme which removes ubiquitin from proteins, leading to the stabilization of SIRT1. This modulation augments the suppression of p53 by SIRT1 while simultaneously increasing mitochondrial biogenesis through PGC-1α, which, in turn, promotes the survival and proliferation of stem cells. This figure was partly created with BioRender.com.
SIRT2. The mitochondrial genome encodes genes essential for the mitochondrial respiratory chain, including OXPHOS subunits, rRNAs, and tRNAs. Due to proximity, mitochondrial oxidative stress may cause mtDNA mutations [
30]. In fact, mtDNA mutations accumulate in stem cells with age and impede the progeny’s mitochondrial oxidative respiration.
SIRT2 is a tubulin deacetylase located in the cytoplasm that has the ability to enter the nucleus during G2–M transition. Its primary function is to regulate the cell cycle. SIRT2 is capable of deacetylating chromatin and microtubules [
22]. The accumulation of mitochondrial stress leads to the activation of the SIRT2-regulated NLRP3 inflammasome [
31]. SIRT2 recognizes NLRP3 as a substrate. SIRT2 deacetylates NLRP3 and thereby inhibits the activation of the NLRP3 inflammasome [
32]. Aged HSCs exhibit a diminished expression of SIRT2, which leads to an abnormal activation of the NLRP3 inflammasome and a heightened vulnerability to stem cell deterioration induced by mitochondrial stress (
Figure 3A) [
31]. SIRT2 is able to delay glycolysis and the metabolic reprogramming of iPSCs by deacetylating four important glycolytic enzymes. These enzymes are phosphoglycerate kinase 1, enolase, aldolase, and GAPDH [
33].
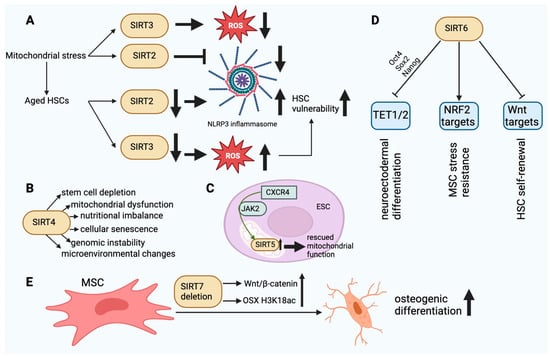
Figure 3. (A). The dysfunction of SIRT2 and SIRT3 in aged HSCs finally leads to HSC vulnerability. In aging HSCs, the activity of SIRT2 is decreased, resulting in an increase in NLRP3 inflammasome activity. In turn, the decrease in SIRT3 activity results in damage to HSCs due to ROS accumulation. (B). The main effects of SIRT4 in SC aging. SIRT4 dysfunction can affect the cellular functions outlined above. (C). SIRT5 upregulation mediated by CXCR4 via JAK2 can rescue normal mitochondrial function in ESCs. (D). SIRT6-mediated epigenetic regulatory functions in SCs. TET1/2 inhibition via Oct4, Sox2, and Nanog may affect neuroectodermal differentiation. Inhibition of Wnt target molecules may regulate the self-renewal capacity of HSCs. Stimulation of NRF2 target molecules may enhance the stress resistance of MSCs. (E). SIRT7 deletion enhances the osteogenic differentiation of MSCs in bone marrow by elevating H3K18ac levels at the promoter of the OSX transcription factor and activating the Wnt/β-catenin signaling pathway. This figure was partly created with BioRender.com.
SIRT3. SIRT3 plays a role in mitochondrial function and metabolism. The translocation of SIRT3 to the nucleus can occur in response to calorie restriction and genotoxic stress. It can deacetylate H4K16 and H3K9 within the nucleus. SIRT3 deacetylates and activates acetyl-CoA synthetase 2 within the mitochondria, thereby augmenting the metabolic rate. In addition, SIRT3 can upregulate the expression of mitochondrial factors such as cytochrome C oxidase subunits, ATP synthetase, and transcription factor PGC1α. SIRT3 can enhance cellular survival by deacetylating Ku70, a protein which is implicated in DNA repair when exposed to genotoxic compounds [
22]. In stem cells, SIRTs have been identified as pivotal regulators of the mitochondrial metabolic checkpoint [
34,
35]. SIRT3 functions as a deacetylase within mitochondria, modifying the antioxidant SOD2 in order to mitigate oxidative stress [
36]. SOD2 expression is significantly upregulated in HSCs, whereas it is downregulated in differentiated hematopoietic cells [
37]. Loss of HSC quiescence and impairment of HSC maintenance and function occur with SIRT3 deletion but not in youth. The functionality of aged HSCs can be enhanced through the overexpression of SIRT3, whereas the expression of the protein is decreased in aging HSCs [
38]. Dysregulation of the mitochondrial metabolic checkpoint is an underlying cause of stem cell senescence and may be amenable to rejuvenation (
Figure 3A).
SIRT4. SIRT4, an ADP-ribosyltransferase located within mitochondria, functions to impede the activity of mitochondrial glutamate dehydrogenase 1. As a result, amino acid-induced insulin secretion is decreased. By deacetylating it, SIRT4 represses the activity of malonyl-CoA decarboxylase. This blocks the oxidation of fatty acids in liver and muscle cells. The inhibitory effect of SIRT4 on PPARα reduces the rate of fatty acid oxidation. Deacetylation of ADP/ATP translocase 2 inhibits mitochondrial uncoupling, thereby increasing cellular ATP [
39]. Stem cell depletion inhibits tissue regeneration and repair [
40,
41]. The gene products of HDAC family members facilitate the differentiation of SCs. The transcriptional downregulation of HDAC can either boost or inhibit SC self-renewal [
42]. SSCs undergo dynamic changes in their transcriptional profiles throughout the processes of self-renewal, differentiation, and aging. SSCs undergoing differentiation and aging exhibits increased expression of SIRT4 and decreased expression of Hdac2, Hdac6, and SIRT1. In response to rapamycin, the expression of HDAC genes changes in SSCs, as shown by lower levels of Hdac8, Hdac9, and SIRT4 transcripts [
43]. The activation of SIRT4 disrupts redox homeostasis and mitochondrial function in trophoblast SCs, which leads to the senescence of stem cells induced by lysine-specific demethylase 1 deficiency [
44]. UV-induced photodamage increases the mRNA expression of SIRT1 and SIRT4 in iPSCs derived from cutaneous fibroblasts. SIRT4 undergoes degradation during the initial phase of photodamage and gradually accumulates during the subsequent phase [
45]. Downregulating SIRT4 with nicotinamide mitigates DNA damage, suggesting that it could be a viable target for preventing somatic cell senescence and promoting iPSC reprogramming [
46]. However, no direct evidence exists at this time to support the notion that SIRT4 is involved in iPSC reprogramming, pluripotency, or differentiation. Contrary to the aforementioned findings, the neural stem cells of adolescent mice exhibited increased SIRT4 expression in comparison to adult mice in a study. In addition, SIRT4 overexpression was found to prevent cell mortality in the presence of DNA damage, indicating that SIRT4 may serve as a protective factor to preserve the genomic integrity of neural stem cells (
Figure 3B) [
47].
The Wnt/β-catenin signaling pathway is essential for the development of numerous organs and the maintenance of SC self-renewal. Mechanisms of degradation and synthesis precisely regulate the maintenance of β-catenin at exceedingly low levels in the cytoplasm in the absence of Wnt ligands. Despite this, the Axin1 protein serves as the primary regulatory target throughout the β-catenin protein degradation pathway. Deacetylation of Axin1, facilitated by SIRT4 translocation from the cytoplasm to the mitochondria and by activation of the Wnt pathway signal, occurs at the K147 residue of Axin1. This deacetylation results in the accumulation of β-catenin protein and a reduction in the β-TrCP assembly of the disruption complex, both of which are consequences of the activation of the Wnt/β-catenin signaling pathway [
48].
SIRT5. Cytochrome C and carbamoyl phosphate synthetase 1 are the targets of SIRT5. SIRT5 demonstrates considerable demalonylase and desuccinylase activity both in vitro and in vivo [
22]. SIRT5 catalyzes the removal of negatively charged modifications, specifically malonyl, succinyl, and glutaryl groups, from the lysine residues of target enzymes, in contrast to SIRT3 and SIRT4 [
49,
50,
51,
52]. Moreover, the role of SIRT5 in metabolic regulation is contingent upon the environment, as particular metabolic processes can be either facilitated or impeded by SIRT5, depending on nutrient availability and cell type [
53,
54,
55,
56].
SIRT5 expression is downregulated in the endothelial progenitor cells of hypertensive patients, resulting in mitochondrial dysfunction characterized by impaired ultrastructure, reduced membrane potential, and increased ROS production [
57,
58]. It was discovered that overexpression of CXCR4 could rescue mitochondrial function in EPC-mediated angiogenesis via JAK2 signaling by upregulating SIRT5 (
Figure 3C) [
58]. MSCs significantly express SIRT5, which contributes to their energy requirements. On the other hand, SIRT5 levels decline considerably in MSCs that have been differentiated from adipose tissue, which is linked to higher levels of succinylation and malonylation in mitochondria [
59]. Consequently, SIRT5 appears to function as an energy demand sensor in these cells. Neuronal SCs express exceptionally high levels of SIRT5, whereas it is significantly reduced in the adult brain. This indicates that SIRT5 expression is elevated in metabolically active stem cell lineages [
47]. SIRT5 deficiency during culture expansion of ADMSCs leads to an abnormal metabolic pattern, increased cell proliferation, and better therapeutic effectiveness [
60]. SIRT5 may be a viable target for enhancing the functional properties of MSCs in preparation for clinical application, according to these findings.
SIRT6. The function of SIRT6 has been associated with gene silencing, DNA repair, and genomic stability. SIRT6 deacetylates the histones H3K9Ac and H3K56, thereby enhancing the stability of telomeres and heterochromatin formation [
22]. SIRT6 is essential for the interaction between epigenetic DNA methylation and the chromatin modifications that occur during ESC differentiation [
61]. SIRT6 inhibits the expression of Oct4, Sox2, and Nanog via the deacetylation of H3K9ac and H3K56ac [
62]. This inhibits the expression of Tet1 and Tet2, which regulate cell lineage selection during ESC differentiation by facilitating 5-hmC of DNA [
63]. Higher amounts of 5-hmC and the overexpression of Tet1 and Tet2 cause neuronal transcription programs to become dysregulated and ESC differentiation to shift toward neuroectodermal lineages in SIRT6-deficient cells [
64].
An increasing body of evidence suggests that SIRT6 plays a crucial role in the regulation of stem cell homeostasis and function across various adult stem cell types. SIRT6 is essential for HSCs’ ability to self-renew over the long term. By deacetylating H3K56, SIRT6 inhibits the transcription of genes involved in the Wnt pathway. The SIRT6 deletion decreased quiescence in HSCs [
65]. SIRT6-dependent histone deacetylation plays a crucial role in MSC homeostasis. In premature or physiological aging, MSC loss may contribute to the breakdown of mesenchymal tissues. Interestingly, SIRT6-deficient rodents display degenerative defects in mesenchymal tissues [
10]. When transplanted in vivo, MSCs deficient in SIRT6 demonstrate impaired differentiation of chondrocytes and osteoblasts, indications of cellular senescence, and an accelerated rate of cellular attrition [
66,
67]. These phenotypes are linked to oxidative stress hypersensitivity, affected redox metabolism, and the downregulation of multiple targets of the NRF2 transcription factor. It is noteworthy that MSCs exhibit a slight elevation in SIRT6 expression as they age. This may serve as a compensatory mechanism to mitigate the detrimental effects of aging-related alterations in MSC functions [
67]. In a past study, SIRT6 overexpression did enhance new bone formation and repair and, by partially inhibiting NFkB signaling, improved osteogenic differentiation of rat MSCs [
68]. SIRT6 plays a crucial function in preserving the epigenetic plasticity of human primary cells prior to their reprogramming into iPSCs. In cells from elderly donors, SIRT6 expression and iPSC reprogramming efficiency are reduced by more than threefold; restoring SIRT6 levels in these cells improves reprogramming effectiveness (
Figure 3D) [
69].
SIRT7. DNA transcription is facilitated by SIRT7 in conjunction with DNA polymerases I–III. In human cells, SIRT7 interacts with two additional molecules: RNA polymerase I and upstream binding factor [
39]. Stem cell exhaustion is a combined effect of different types of damage related to aging and is likely a main cause of the breakdown of body tissues and cells [
70], hindering their regenerative capacity. In SIRT7-deficient mice, there is an increase in HSCs in the bone marrow. However, these cells display a diminished capacity to repopulate the lymphoid compartment of lethally irradiated mice compared to wild-type bone marrow cells [
71]. The reason for this is that the inhibition of transcription by SIRT7 of mitochondrial ribosome components is crucial in preventing HSCs from losing their quiescence, which would otherwise lead to proliferation and a decrease in survival [
72,
73]. In hair follicle stem cells, SIRT7 keeps telogen quiescence going. This phenomenon can be elucidated by the mechanism by which SIRT7 deacetylates residue K612 of NFATc1, thereby promoting its phosphorylation by GSK-3β, which subsequently inhibits the protein’s signaling and leads to its nuclear degradation via PA28c [
74]. SIRT7 deficiency in human MSCs is associated with the emergence of various senescence-related characteristics, including retrotransposons such as LINE1 and accelerated functional wasting caused by the decondensation of heterochromatin in the nuclear periphery and activation of the cGAS-STING pathway [
75]. In the context of intestinal epithelial homeostasis, SIRT7 plays a critical role, particularly in the maintenance of LGR5+ stem cells which are essential for epithelial regeneration. A deficiency in this protein results in aberrant chromosome segregation, an insufficient response to DNA damage, and insufficient intestinal cell differentiation. Furthermore, it increases Wnt signaling and the expression of genes associated with colorectal cancer [
76]. MiR-152 has been identified to increase with the aging of human DPSCs and to promote the senescent phenotype via its regulatory effect on SIRT7 levels [
77]. By increasing the H3K18ac levels at the promoter of the OSX transcription factor and by activating the Wnt/β-catenin signaling pathway, SIRT7 deletion promotes the osteogenic differentiation of mesenchymal stem cells in bone marrow (
Figure 3E) [
78,
79]. This is particularly true for isoforms 1 and 2. RBM6 guides the binding of SIRT7 to the OSX promoter, which is governed by the lncRNA PLXDC2-OT [
79]. Additionally, a protracted in vitro culture of iPSCs resulted in the detection of low levels of SIRT7. These results replicate various aspects related to premature aging syndromes and somatic cell senescence, as documented by previous studies [
80]. SIRT7-mediated maintenance of stem cell quiescence is critical for population preservation. However, it can disrupt processes which require stem cells to differentiate, necessitating appropriate regulation. Exosomes secreted by bone marrow MSCs carrying miR-125b downregulate SIRT7, enhancing the response to myocardial ischemia-reperfusion injury [
81]. This may be achieved by facilitating the replacement of damaged cells in a more timely and effective manner. In the same way, exosomal miR-17-5p, which comes from human umbilical cord mesenchymal stem cells, lowers SIRT7 to improve ovarian function in women who have already lost their ovaries [
82]. Overexpression of SIRT7 in mouse embryonic fibroblasts has been shown to inhibit cell proliferation and growth by activating transcription in the absence of p53 and c-Myc [
83].
In conclusion, it is evident that SIRTs serve a critical regulatory function in SCs. The SIRT family is known for its roles in metabolic diseases and aging. They are also important regulators of SC biology, acting as stress and metabolic sensors in cells. Diverse SIRTs exhibit cell type-specific and/or stage-dependent effects on stem cells in response to environmental stimuli, regardless of their shared reliance on intracellular NAD+. Nuclear and cytosolic SIRTs have an effect on pluripotent stem cells through epigenetics, redox homeostasis, controlling metabolism and the stress response of cells, and managing the factors that determine pluripotency. SIRTs safeguard against the stress-induced and aging-induced depletion of adult stem cells by preserving their capacity for self-renewal, quiescence, and regeneration.
Regarding carcinogenesis, SIRT1, SIRT3, SIRT4, and SIRT6 operate similarly in healthy cells to tumor suppressor proteins. They independently advocate for the maintenance of genomic stability and DDR. SIRT3 and SIRT6 are conceivably the finest examples of how numerous SIRTs activate “real” tumor suppressor proteins, such as gatekeepers and caretakers, while inactivating oncoproteins. In these particular circumstances, the mere presence of upregulated SIRTs in certain cancer cells does not necessarily imply that they play a causal role in the development of cancer [
84]. However, impaired SIRT functions can lead to DNA damage, genomic instability, and chromosomal aberrations, which can be the basis of tumorigenesis [
84].
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines12020386